| Acrocephalosyndactyly | |
|---|---|
| Other names | ACS |
 | |
| Syndactyly in acrocephalosyndactyly (Apert) | |
| Specialty | Medical genetics |
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly).[1] Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth.[2][3] Syndactyly occurs when digits of the hands or feet are fused together.[4] When polydactyly is also present, the classification is acrocephalopolysyndactyly.[5] Polydactyly occurs when the hands or feet possess additional digits.[6] Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern[1] Treatment often involves surgery in early childhood to correct for craniosynostosis[7] and syndactyly.[8]
The severity of symptoms for acrocephalosyndactyly varies significantly by subtype and treatment in the early stages of life.
History
Cases of the condition have been recorded as early as the 18th century.[9] The term acrocephalosyndactyly (from Greek ἄκρος (ákros) ‘highest, at the extremity’, κεφαλή (kephalḗ) ‘head’, σύν (syn) 'together' and δάκτυλος (daktylos) 'finger') was first applied in 1906 by French physician Eugène Apert first to describe a condition characterized by craniosynostosis and syndactyly.[10] The condition described by Apert is now known as the Apert syndrome subtype of acrocephalosyndactyly.[11] Other subtypes of acrocephalosyndactyly were characterized throughout the 20th century.[1]
Prevalence
Considering all types of acrocephalosyndactyly, one newborn baby is born with acrocephalosyndactyly for every 65,000 - 102,500 babies born. There is no difference in the amount of males and females affected by acrocephalosyndactyly.[12][13][14][15]
Characteristics
Acrocephalosyndactyly presents in numerous different subtypes, however, considerable overlap in symptoms occurs. Generally, all forms of acrocephalosyndactyly are characterized by atypical craniofacial, hand, and foot characteristics, such as premature closure of the fibrous joints in between certain bones of the skull,[16][17] fusion of certain fingers or toes,[16][18] and/or more than the usual number of digits.[19] Some subtypes also involve structural heart variations that are present at birth.[20][1]
Cause
Most forms of acrocephalosyndactyly or acrocephalopolysyndactyly are inherited in autosomal dominant pattern,[15][20][1] with the exclusion of Carpenter Syndrome which is inherited in autosomal recessive manner. De-novo variants, or genetic alterations not inherited from one's parents, in different genes were reported to cause several types of acrocephalosyndactyly. Among known genetic changes, there are variations in genes such as Fibroblast growth factor receptor (FGFR),[16][17][21][22] TWIST1,[23] and RAB23.[24] Constitutive activation in these categories of genes, particularly FGFR, which is heavily involved during the development stage of embryos such as organ development or organogenesis and the maintenance of tissue forming cells, known as progenitor cells, can be very detrimental.[25]
Genetically inherited acrocephalosyndactyly conditions all show high to complete penetrance with a variable expression, meaning that all individuals who inherit the condition present atypical characteristic craniofacial, hand, and foot structures, but the severity of disabilities is variable.[1] Increased paternal age is considered a risk factor in some cases.[1]
Impacts of Conditions on Life
Despite the current major efforts of surgical therapeutics on the effects of Acrocephalosyndactyly, morbidities still exist within individuals that have received treatment. Those who reach adulthood often have lower levels of education than their peers, as well as greater difficulty in various social aspects, such as dating, marriage, or sexual relationships. They may also report the need for assisted living throughout their life as well as other health issues, such as hearing issues or epilepsy at a more common frequency than their counterparts.[26]
Fortunately, many individual with the condition report similar levels of happiness with their lives as non-afflicted individuals[26] and show high social integration as well as great physical and emotional resilience despite any impediments.[27]
Diagnosis
Prenatal Diagnosis
Diagnosis prior to birth is possible for some forms of acrocephalosyndactyly. Prenatal genetic diagnosis is only possible if the gene variation responsible for the syndrome is known and the variation causing the disease has been identified within the genome of a family member. Collection of samples for genetic testing can be done using amniocentesis, which samples embryonic stem cells contained in amniotic fluid, or chorionic villus sampling, which samples placental cells.[15] There has been a case of a prenatal diagnosis of Apert syndrome using fetoscopy, where the fetus is observed using an endoscope inserted into the uterus from the abdomen.[22] Alternatively, there has been interest in using non-invasive techniques like ultrasound to detect atypical fetal skull features.[28]
Postnatal Diagnosis
Most diagnoses of acrocephalosyndactyly occur after birth by assessing the physical symptoms of the infant. This can be supported with radiographic imaging, such as X-ray imaging, and molecular genetic testing, which looks for DNA variations known to cause the disease.[29][30] Molecular genetic testing typically occurs in the FGFR, TWIST1, and RAB23 genes.
Nomenclature/Classification
There is no consistent nomenclature or classification across the different syndromes under the umbrella of acrocephalosyndactyly and acrocephalopolysyndactyly. Although acrocephalosyndactyly has been reported as early as the 18th century,[9] the ACS and ACPS classifications only came in the latter 20th century. However, this classification may be outdated as it has been suggested that the distinction between acrocephalosyndactyly and acrocephalopolysyndactyly should be erased.[5]
Currently, Noack syndrome (ACPS type I) is now classified as Pfeiffer syndrome (ACS type V);[31] Goodman syndrome (ACPS type IV) is classified as a variation of Carpenter syndrome (ACPS type II);[19] and different researchers have combined Apert (ASC type I), Crouzon (ASC type II), and Pfeiffer (ASC type V) syndrome into Apert-Crouzon[32] and Crouzon-Pfeiffer[33] syndrome.
Acrocephalosyndactyly type IV was formerly called Mohr Syndrome, however, it was later classified under Orofaciodigital syndrome type II.[34] Pfeiffer syndrome was formerly type VI and Waardenburg type V, but this was changed sometime after 1966.[35]
Acrocephalosyndactyly (ACS):
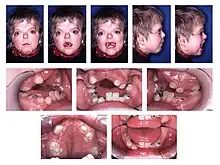
- type I – Apert syndrome[11]
- type II – Crouzon syndrome[36]
- type III – Saethre–Chotzen syndrome[37]
- Robinow-Sorauf syndrome suggested to be included in Saethre-Chotzen classification [38]
- type IV – Mohr syndrome (archaic) [34][35]
- type V – Pfeiffer syndrome[31]
- Noack syndrome incorporated into Pfeiffer syndrome classification [31]
.jpg.webp)
A related term, acrocephalopolysyndactyly (ACPS), refers to the inclusion of polydactyly to the presentation. It also has multiple types:
- type I – Noack syndrome (archaic) [31]
- type II – Carpenter syndrome[39]
- Goodman syndrome incorporated into Carpenter syndrome classification [40]
- Summitt syndrome incorporated into Carpenter syndrome classification [41]
- type III – Sakati-Nyhan-Tisdale syndrome[42]
- type IV – Goodman syndrome (archaic) [40]
Treatment
Craniosynostosis
For subtypes with craniosynostosis, surgery is required to prevent premature fusion of cranial sutures, such as the coronal suture (brachycephaly).[7] The cranial suture located between the two frontal and two parietal skull bones is called the coronal suture.[2] Cranioplasty should be performed in the first year of life to prevent disruptions in brain growth due to increased intracranial pressure. Midface surgery may also be required in childhood to detach the midface from the rest of the skull to correct respiratory and orthodontic problems.[43][44]
Syndactyly
Syndactyly in certain subtypes is rarely severe enough to affect hand function, so treatment may not be needed.[43]
In more severe subtypes, as seen in Apert syndrome, surgical correction of syndactyly may be needed. Surgery is recommended to be performed as soon as possible, generally at 4 months of age. Treatment is dependent on the severity of syndactyly. The surgical treatment generally involves interdigital webspace release and thumb lengthening.[8]
Management
Treatment for those diagnosed with acrocephalosyndactyly extends beyond surgery. There are many steps that can aid in long-term management of the syndrome. Individuals afflicted with acrocephalosyndactyly and their caregivers can build a health care support system by building strong relationships with a team of medical specialists. Preformed teams of medical specialists can often be found at universities or research institutions. Caregivers can prevent future challenges by exploring options for financial aid, health insurance, and accommodating educational institutions. Primary caregivers are encouraged to prioritize their emotional health by reserving time for themselves and by sourcing a reliable support system.[29]
See also
References
- 1 2 3 4 5 6 7 Bissonnette, Bruno; Luginbuehl, Igor; Marciniak, Bruno; Dalens, Bernard J. (2006), "Acrocephalosyndactyly Syndromes", Syndromes: Rapid Recognition and Perioperative Implications, New York, NY: The McGraw-Hill Companies
- 1 2 Russell, William P.; Russell, Mark R. (2023), "Anatomy, Head and Neck, Coronal Suture", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30252267, retrieved 4 December 2023
- ↑ Opperman, L. A. (2000). "Cranial sutures as intramembranous bone growth sites". Developmental Dynamics. 219 (4): 472–485. doi:10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. ISSN 1058-8388. PMID 11084647. S2CID 8801611.
- ↑ Malik, S. (2012). "Syndactyly: phenotypes, genetics and current classification". European Journal of Human Genetics. 20 (8): 817–824. doi:10.1038/ejhg.2012.14. ISSN 1476-5438. PMC 3400728. PMID 22333904.
- 1 2 Cohen, M. Michael; Kreiborg, Sven (May 1995). "Hands and feet in the Apert syndrome". American Journal of Medical Genetics. 57 (1): 82–96. doi:10.1002/ajmg.1320570119. ISSN 0148-7299. PMID 7645606.
- ↑ Malik, S. (2014). "Polydactyly: phenotypes, genetics and classification". Clinical Genetics. 85 (3): 203–212. doi:10.1111/cge.12276. ISSN 0009-9163. PMID 24020795. S2CID 22412404.
- 1 2 Craniosynostosis : diagnosis, evaluation, and management. M. Michael Cohen, Ruth E. MacLean (2nd ed.). New York: Oxford University Press. 2000. ISBN 0-19-511843-X. OCLC 41528658.
{{cite book}}: CS1 maint: others (link) - 1 2 Raposo-Amaral, Cassio Eduardo; Denadai, Rafael; Furlan, Pedro; Raposo-Amaral, Cesar Augusto (October 2018). "Treatment of Apert Hand Syndrome: Strategies for Achieving a Five-Digit Hand". Plastic and Reconstructive Surgery. 142 (4): 972–982. doi:10.1097/PRS.0000000000004815. ISSN 0032-1052. PMID 29994846. S2CID 51614940.
- 1 2 Wheaton, S. W. (1894). "Two specimens of congenital cranial deformity in infants associated with fusion of the fingers and toes". Trans Pathol Soc Lond. 45: 238–241.
- ↑ Apert, M. E. (1906). "De l'acrocephalosyndactylie". Bull. Mem. Soc. Med. Hop. Paris. 23: 1310–1330.
- 1 2 "Entry - #101200 - Apert Syndrome". omim.org. Retrieved 4 December 2023.
- ↑ M Das, Joe; Winters, Ryan (2023), "Pfeiffer Syndrome", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30422477, retrieved 4 December 2023
- ↑ Conrady, Christopher D.; Patel, Bhupendra C.; Sharma, Sandeep (2023), "Apert Syndrome", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30085535, retrieved 4 December 2023
- ↑ Tolarova, M. M. (2023). "Genetics of Crouzon Syndrome". Pediatrics: Genetics and Metabolic Disease – via Medscape.
- 1 2 3 Gallagher, Emily R.; Ratisoontorn, Chootima; Cunningham, Michael L. (1993), Adam, Margaret P.; Feldman, Jerry; Mirzaa, Ghayda M.; Pagon, Roberta A. (eds.), "Saethre-Chotzen Syndrome", GeneReviews®, Seattle (WA): University of Washington, Seattle, PMID 20301368, retrieved 4 December 2023
- 1 2 3 Wilkie, Andrew O. M.; Slaney, Sarah F.; Oldridge, Michael; Poole, Michael D.; Ashworth, Geraldine J.; Hockley, Anthony D.; Hayward, Richard D.; David, David J.; Pulleyn, Louise J.; Rutland, Paul; Malcolm, Susan; Winter, Robin M.; Reardon, William (February 1995). "Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome". Nature Genetics. 9 (2): 165–172. doi:10.1038/ng0295-165. ISSN 1546-1718. PMID 7719344. S2CID 12423131.
- 1 2 Carinci, Francesco; Pezzetti, Furio; Locci, Paola; Becchetti, Ennio; Carls, Friedrick; Avantaggiato, Anna; Becchetti, Alessio; Carinci, Paolo; Baroni, Tiziano; Bodo, Maria (May 2005). "Apert and Crouzon Syndromes: Clinical Findings, Genes and Extracellular Matrix". Journal of Craniofacial Surgery. 16 (3): 361–368. doi:10.1097/01.SCS.0000157078.53871.11. ISSN 1049-2275. PMID 15915098. S2CID 23327865.
- ↑ Bissonnette, Bruno; Luginbuehl, Igor; Engelhardt, Thomas (2019), "Acrocephalopolysyndactyly Type IV: Goodman Syndrome", Syndromes: Rapid Recognition and Perioperative Implications (2 ed.), New York, NY: McGraw-Hill Education
- 1 2 Cohen, Donald M.; Green, James G.; Miller, Janice; Gorlin, Robert J.; Reed, Jerry A.; Opitz, John M.; Reynolds, James F. (October 1987). "Acrocephalopolysyndactyly type II—Carpenter syndrome: Clinical spectrum and an attempt at unification with Goodman and Summit syndromes". American Journal of Medical Genetics. 28 (2): 311–324. doi:10.1002/ajmg.1320280208. ISSN 0148-7299. PMID 3322002.
- 1 2 Kumar, Niraj; Arora, Shubhangi; Bindra, Ashish; Goyal, Keshav (2014). "Anesthetic management of craniosynostosis repair in patient with Apert syndrome". Saudi Journal of Anaesthesia. 8 (3): 399–401. doi:10.4103/1658-354X.136631. ISSN 1658-354X. PMC 4141395. PMID 25191197.
- ↑ Galvin, B D; Hart, K C; Meyer, A N; Webster, M K; Donoghue, D J (23 July 1996). "Constitutive receptor activation by Crouzon syndrome mutations in fibroblast growth factor receptor (FGFR)2 and FGFR2/Neu chimeras". Proceedings of the National Academy of Sciences. 93 (15): 7894–7899. Bibcode:1996PNAS...93.7894G. doi:10.1073/pnas.93.15.7894. ISSN 0027-8424. PMC 38845. PMID 8755573.
- 1 2 Leonard, Claire O.; Daikoku, Norman H.; Winn, Kevin; Opitz, John M. (January 1982). "Prenatal fetoscopic diagnosis of the Apert syndrome". American Journal of Medical Genetics. 11 (1): 5–9. doi:10.1002/ajmg.1320110103. ISSN 0148-7299. PMID 7065003.
- ↑ Cai, Juanliang; Goodman, Barbara K.; Patel, Ankita S.; Mulliken, John B.; Van Maldergem, Lionel; Hoganson, George E.; Paznekas, William A.; Ben-Neriah, Ziva; Sheffer, Ruth; Cunningham, Michael L.; Daentl, Donna L.; Jabs, Ethylin Wang (1 December 2003). "Increased risk for developmental delay in Saethre-Chotzen syndrome is associated with TWIST deletions: an improved strategy for TWIST mutation screening". Human Genetics. 114 (1): 68–76. doi:10.1007/s00439-003-1012-7. ISSN 1432-1203. PMID 14513358. S2CID 20929600.
- ↑ Jenkins, Dagan; Seelow, Dominik; Jehee, Fernanda S.; Perlyn, Chad A.; Alonso, Luís G.; Bueno, Daniela F.; Donnai, Dian; Josifiova, Dragana; Mathijssen, Irene M. J.; Morton, Jenny E. V.; Ørstavik, Karen Helene; Sweeney, Elizabeth; Wall, Steven A.; Marsh, Jeffrey L.; Nürnberg, Peter (1 June 2007). "RAB23 Mutations in Carpenter Syndrome Imply an Unexpected Role for Hedgehog Signaling in Cranial-Suture Development and Obesity". The American Journal of Human Genetics. 80 (6): 1162–1170. doi:10.1086/518047. ISSN 0002-9297. PMC 1867103. PMID 17503333.
- ↑ Yamaji, Kojiro; Morita, Jumpei; Watanabe, Tsukasa; Gunjigake, Kaori; Nakatomi, Mitsushiro; Shiga, Momotoshi; Ono, Kentaro; Moriyama, Keiji; Kawamoto, Tatsuo (November 2018). "Maldevelopment of the submandibular gland in a mouse model of apert syndrome". Developmental Dynamics. 247 (11): 1175–1185. doi:10.1002/dvdy.24673. ISSN 1058-8388. PMID 30251381. S2CID 52815441.
- 1 2 Tovetjärn, Robert; Tarnow, Peter; Maltese, Giovanni; Fischer, Sara; Sahlin, Per-Erik; Kölby, Lars (October 2012). "Children with Apert Syndrome as Adults: A Follow-Up Study of 28 Scandinavian Patients". Plastic & Reconstructive Surgery. 130 (4): 572e–576e. doi:10.1097/PRS.0b013e318262f355. ISSN 0032-1052. PMID 23018718. S2CID 45847015.
- ↑ Taghinia, Amir H.; Yorlets, Rachel R.; Doyle, Michael; Labow, Brian I.; Upton, Joseph (April 2019). "Long-Term Functional Upper-Extremity Outcomes in Adults with Apert Syndrome". Plastic & Reconstructive Surgery. 143 (4): 1136–1145. doi:10.1097/PRS.0000000000005479. ISSN 0032-1052. PMID 30676503. S2CID 59225959.
- ↑ "The encyclopedia of genetic disorders and birth defects | WorldCat.org". search.worldcat.org. Retrieved 4 December 2023.
- 1 2 "Apert syndrome - Diagnosis & Treatment - Genetic and Rare Diseases Information Center". rarediseases.info.nih.gov. Retrieved 5 November 2022.
- ↑ Gonzales, M.; Heuertz, S.; Martinovic, J.; Delahaye, S.; Bazin, A.; Loget, P.; Pasquier, L.; Le Merrer, M.; Bonaventure, J. (17 June 2005). "Vertebral anomalies and cartilaginous tracheal sleeve in three patients with Pfeiffer syndrome carrying the S351C FGFR2 mutation: Letter to the Editor". Clinical Genetics. 68 (2): 179–181. doi:10.1111/j.1399-0004.2005.00477.x. PMID 15996217. S2CID 1652216.
- 1 2 3 4 "Entry #101600 - Pfeiffer Syndrome". omim.org. Retrieved 4 December 2023.
- ↑ Rédei, George P., ed. (2008), "Apert or Apert-Crouzon Syndrome", Encyclopedia of Genetics, Genomics, Proteomics and Informatics, Dordrecht: Springer Netherlands, p. 127, doi:10.1007/978-1-4020-6754-9_1009, ISBN 978-1-4020-6754-9, retrieved 5 November 2022
- ↑ Wilson, Alexander T.; de Planque, Catherine A.; Yang, Sumin S.; Tasker, Robert C.; van Veelen, Marie-Lise C.; Dremmen, Marjolein H. G.; Vrooman, Henri A.; Mathijssen, Irene M. J. (October 2020). "Cortical Thickness in Crouzon-Pfeiffer Syndrome: Findings in Relation to Primary Cranial Vault Expansion". Plastic and Reconstructive Surgery. Global Open. 8 (10): e3204. doi:10.1097/GOX.0000000000003204. ISSN 2169-7574. PMC 7647527. PMID 33173703.
- 1 2 "Entry - %252100 - Mohr Syndrome". omim.org. Retrieved 5 November 2022.
- 1 2 McKusick, V. A. (1966). "Autosomal Dominant Phenotypes". Mendelian inheritance in man: Catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes. Johns Hopkins Press. pp. 3–5.
- ↑ "Entry - #123500 - CROUZON SYNDROME - OMIM". omim.org. Retrieved 5 November 2022.
- ↑ "Entry - #101400 - SAETHRE-CHOTZEN SYNDROME; SCS - OMIM". omim.org. Retrieved 5 November 2022.
- ↑ Reardon, W.; Winter, R. M. (1994). "Saethre-Chotzen syndrome". Journal of Medical Genetics. 31 (5): 393–396. doi:10.1136/jmg.31.5.393. ISSN 0022-2593. PMC 1049872. PMID 8064818.
- ↑ "Entry #201000 - Carpenter Syndrome 1; CRPT1". omim.org. Retrieved 4 December 2023.
- 1 2 "Entry 201020 - Acrocephalopolysyndactyly type IV". omim.org. Retrieved 4 December 2023.
- ↑ "Entry 272350 - Summitt Syndrome". omim.org. Retrieved 4 December 2023.
- ↑ "Entry 101120 - Acrocephalopolysyndactyly type III". omim.org. Retrieved 4 December 2023.
- 1 2 Anderson, Peter. "Headlines Craniofacial Support" (PDF). Archived from the original on 30 June 2012.
- ↑ "Pfeiffer Syndrome". NORD (National Organization for Rare Disorders). Retrieved 5 November 2022.
External links
- Acrocephalosyndactylia at the U.S. National Library of Medicine Medical Subject Headings (MeSH)