The α-ketol rearrangement is the acid-, base-, or heat-induced 1,2-migration of an alkyl or aryl group in an α-hydroxy ketone or aldehyde to give an isomeric product.[1]
Introduction
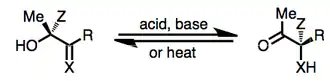
Like other ketogenic rearrangements, the α-ketol rearrangement involves the transformation of an alkoxide into a carbonyl group with concomitant movement of the bonding electrons of the migrating group towards an adjacent trigonal center. A distinctive feature of this particular rearrangement, however, is its reversibility—as a result, the more stable α-hydroxy carbonyl compound is favored. A general scheme for the rearrangement is shown below.
(1)
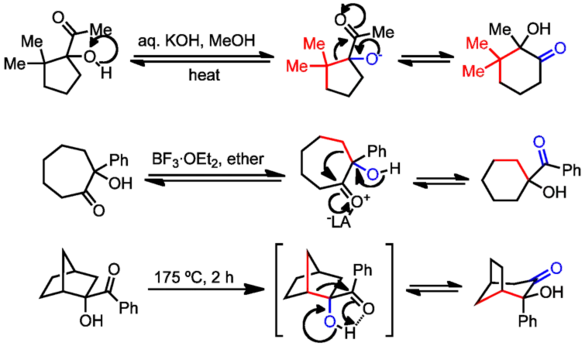
This rearrangement differs from similar isomerizations of carbohydrates, which involve the migration of hydrogen and proceed through discrete enediol intermediates. These include the Lobry–de Bruyn–van Ekenstein transformation,[2] the Heyns[3] and Amadori rearrangements,[4] and the Voight[5] and Bilik[6] reactions. α-hydroxy imines may also undergo the rearrangement, although the thermodynamic driving force to amino ketones is often weak (in the absence of protic acids; see below).
Advantages: Large thermodynamic energy differences between reactants and products can be harnessed to drive these reactions to completion. Reaction progress can also be influenced through conformational control and often exhibit asymmetric induction.
Disadvantages: Because the reaction is reversible and thermodynamically controlled, it cannot be used to synthesize unstable α-hydroxy carbonyl products. Ideal conditions are often difficult to pinpoint and can require extensive catalyst screening.
Mechanism and stereochemistry
Prevailing mechanism
Under basic conditions, the reaction is initiated by deprotonation of the hydroxyl group.[7] Substrates must lack α-hydrogens to prevent competitive reactions involving enolates. Under Brønsted- or Lewis-acidic conditions, coordination to the carbonyl oxygen occurs first, and under thermal conditions, intramolecular proton transfer takes place at the same time as migration. The reversibility of the reaction implies that reaction products are more thermodynamically stable than the corresponding starting materials. Starting materials incorporating ring strain, for instance, will rearrange to products lacking strain.
(2)
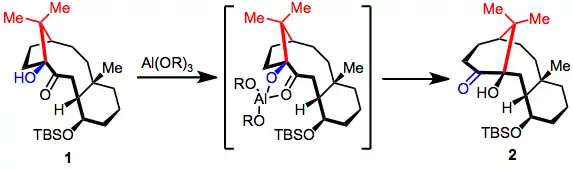
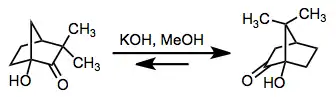
When metal salts are used to promote rearrangement, stereoelectronic effects derived from chelation to the metal salt can enhance the speed and selectivity of the reaction. In the conversion of 1 to 2, for instance, coordination of both the hydroxyl and carbonyl groups to aluminum facilitates rapid, selective migration of the bond to the one-carbon bridge.[8] Similar stereoelectronic effects were observed in studies of the rearrangement of 17-hydroxy-20-ketosteroids. In this case, Lewis-acidic conditions switched the sense of stereoselectivity observed for the base-catalyzed process.
(3)
α-Hydroxy imines may also undergo rearrangement to amino ketones. Hammett analysis and a very negative entropy of activation suggest that the reaction proceeds in a single step through a concerted transition state.[9] As a result, subtle conformational and steric factors can play a role in the speed and extent of these reactions. Allylic transposition has been observed in migrations of allyl groups, but propargyl groups undergo simple alkyl migration.[10]
Enantioselective variants
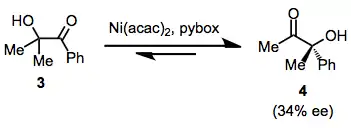
Although examples of enantioselective α-ketol rearrangements starting from achiral α-hydroxy ketones are fairly limited, a number of examples of 1,2-asymmetric induction (due to stereoelectronic factors) have been observed. In one example of an enantioselective process, use of nickel(II) diacetoacetonate and pybox provided 4 in 34% ee.[11]
(4)
If the relative orientation of the carbonyl and hydroxy group can be controlled (through intramolecular hydrogen bonding, for instance), stereoselectivity can be achieved. This conformational control forces the migrating group to form its new bond to a single face of the carbonyl group.
Scope and limitations
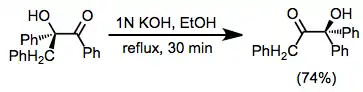
The scope of rearrangements of α-hydroxy ketones and aldehydes is limited only by the fact that the product must be more thermodynamically stable than the starting material. In some cases, very subtle structural differences dictate the favored direction of isomerization. For instance, according to the "Favorskii rule," an empirical guideline with numerous exceptions, products with the carbonyl group adjacent to a methyl group or distal to a phenyl group are favored over the corresponding isomers.[12] In many subtle cases, such as the one below, decreased nonbonding interactions in dominant conformations of the favored isomers are often invoked[13]
(5)
Alkoxyallenes with an α-hydroxy substituent may provide allylic alcohols after rearrangement. Ring expansion provides the thermodynamic driving force in this case.[14]
(6)
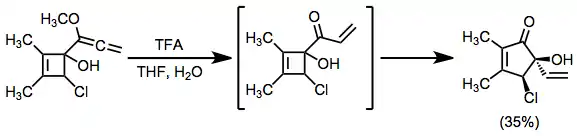
Steroidal ketols have been subjected to rearrangement conditions to give steroids of different ring sizes. These rearrangements often proceed with a high degree of stereocontrol.[15]
(7)
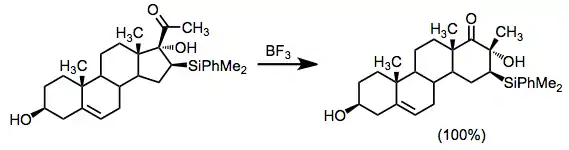
Bridged ketols also undergo rearrangement, often stereospecifically.[16]
(8)
α-Hydroxy aldehydes have a strong thermodynamic preference for rearrangement to the corresponding ketols in the absence of steric or other factors.
Rearrangements of α-hydroxy imines are more difficult to predict because of the small energy differences between isomers. One synthetically useful application of this rearrangement is to the synthesis of spirocycles: fused hydroxyimines can rearrange to give the corresponding spiro isomers.[17]
(9)
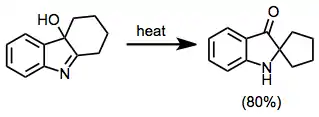
Comparison with other methods
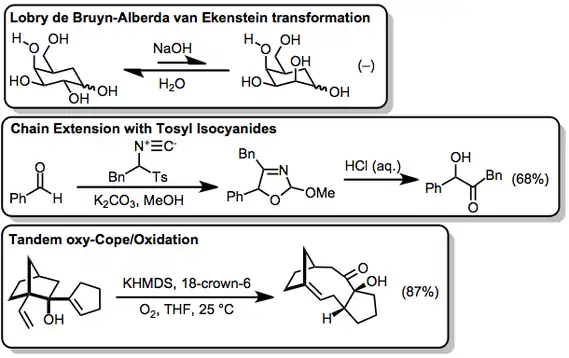
Ketol isomerizations of secondary α-hydroxy carbonyl compounds, which proceed via tautomerization, are probably the closest relative of the α-ketol rearrangement discussed here. Also closely related are carbohydrate rearrangements such as the Lobry–de Bruyn–van Ekenstein transformation,[18] which involve conversion to the open form followed by rearrangement and re-closing.
Synthesis of α-hydroxy carbonyl compounds is most commonly accomplished by either chain extension or oxidation of carbonyl compounds. In conjunction with aldehydes and ketones, tosyl isocyanides can be used to form α-hydroxy ketones after hydrolysis of the resulting oxazolines. The sp-hybridized isocyanide carbon becomes the carbonyl carbon of the product.[19] A variety of reagents exist to oxidize enolates to α-hydroxy carbonyl compounds—in the example here, oxy-Cope rearrangement generates an enolate, which is then oxidized in the presence of molecular oxygen.[20]
(10)
Experimental conditions and procedure
Typical conditions
The most common experimental procedures for the rearrangement of α-ketols involve simple heating or exposure to a base or an acid. However, discovering the ideal conditions for the reaction often requires extensive optimization—simple Bronsted acids and bases do not always work well. Group 13 Lewis acids have been shown to work well as catalysts; however coordination of the catalyst has important stereoelectronic consequences. Additionally, under thermal conditions, intramolecular hydrogen bonding may influence the product distribution. Conditions for the rearrangement of α-hydroxy imines are similar, although the resulting amino ketone products are usually isolated as the corresponding acid salts.
References
- ↑ Paquette, L. A.; Hofferberth, J. E. Org. React. 2003, 62, 477–567. doi:10.1002/0471264180.or062.03
- ↑ Gottfried, J.; Benjamin, G. Ind. Eng. Chem. 1952, 44, 141.
- ↑ Wrodnigg, T. M.; Eder, B. Top. Curr. Chem. 2001, 215, 115.
- ↑ Hodge, J. Adv. Carbohydr. Chem. 1955, 10, 169.
- ↑ Voight, K. J. Prakt. Chem. 1886, 34, 1. DOI: 10.1002/prac.18860340101
- ↑ Petrus, L.; Petrusova, M.; Hricoviniova, Z. Top. Curr. Chem. 2001, 215, 15.
- ↑ Gelin, S.; Gelin, R. J. Org. Chem. 1979, 44, 808.
- ↑ Paquette, L. A.; Montgomery, F. J.; Wang, T. Z. J. Org. Chem. 1995, 60, 7857.
- ↑ Stevens, C. L.; Hanson, H. T.; Taylor, K. G. J. Am. Chem. Soc. 1966, 88, 2769.
- ↑ Vatèle, J.-M.; Dumas, D.; Goré, J. Tetrahedron Lett. 1990, 31, 2277.
- ↑ Brunner, H.; Stöhr, F. Eur. J. Org. Chem. 2000, 2777.
- ↑ Colard, P.; Elphimoff-Felkin, I.; Verrier, M. Bull. Soc. Chim. Fr. 1961, 516.
- ↑ Brunner, H.; Stöhr, F. Eur. J. Org. Chem. 2000, 2777.
- ↑ Paukstelis, J. V.; Kao, J.-1. J. Am. Chem. Soc. 1972, 94, 4783.
- ↑ Bischofberger, N.; Walker, K. A. M. J. Org. Chem. 1985, 50, 3604.
- ↑ Creary, X.; Inocencio, P. A.; Underiner, T. L.; Kostromin, R. J. Org. Chem. 1985, 50, 1932.
- ↑ Witkop, B.; Patrick, J. B. J. Am. Chem. Soc. 1951, 73, 2188.
- ↑ Angyal, S. J. Top. Curr. Chem., 2001, 215, 1.
- ↑ Van Leusen, D.; van Leusen, A. M. Org. React. 2001, 57, 417.
- ↑ Paquette, L. A.; DeRussy, D. T.; Pegg, N. A.; Taylor, R. T.; Zydowsky, T. M. J. Org. Chem. 1989, 54, 4576.