| Porphyria | |
|---|---|
 | |
| Left figure is urine on the first day while the right figure is urine after three days of sun exposure showing the classic change in color to purple. | |
| Pronunciation |
|
| Specialty | Hematology, dermatology, neurology |
| Symptoms | Depending on subtype—abdominal pain, chest pain, vomiting, confusion, constipation, fever, seizures, blisters with sunlight[1][2] |
| Usual onset | Recurrent attacks that last days to weeks[2] |
| Causes | Usually genetic[2] |
| Diagnostic method | Blood, urine, and stool tests, genetic testing[2] |
| Differential diagnosis | Lead poisoning, alcoholic liver disease[3] |
| Treatment | Depends on type and symptoms[2] |
| Frequency | 1 to 100 in 50,000 people[1] |
Porphyria is a group of liver disorders in which substances called porphyrins build up in the body, adversely affecting the skin or nervous system.[1] The types that affect the nervous system are also known as acute porphyria, as symptoms are rapid in onset and short in duration.[1] Symptoms of an attack include abdominal pain, chest pain, vomiting, confusion, constipation, fever, high blood pressure, and high heart rate.[1][2][4] The attacks usually last for days to weeks.[2] Complications may include paralysis, low blood sodium levels, and seizures.[4] Attacks may be triggered by alcohol, smoking, hormonal changes, fasting, stress, or certain medications.[2][4] If the skin is affected, blisters or itching may occur with sunlight exposure.[2]
Most types of porphyria are inherited from one or both of a person's parents and are due to a mutation in one of the genes that make heme.[2] They may be inherited in an autosomal dominant, autosomal recessive, or X-linked dominant manner.[1] One type, porphyria cutanea tarda, may also be due to hemochromatosis (increased iron in the liver), hepatitis C, alcohol, or HIV/AIDS.[1] The underlying mechanism results in a decrease in the amount of heme produced and a build-up of substances involved in making heme.[1] Porphyrias may also be classified by whether the liver or bone marrow is affected.[1] Diagnosis is typically made by blood, urine, and stool tests.[2] Genetic testing may be done to determine the specific mutation.[2]
Treatment depends on the type of porphyria and the person's symptoms.[2] Treatment of porphyria of the skin generally involves the avoidance of sunlight, while treatment for acute porphyria may involve giving intravenous heme or a glucose solution.[2] Rarely, a liver transplant may be carried out.[2]
The precise prevalence of porphyria is unclear, but it is estimated to affect between 1 and 100 per 50,000 people.[1] Rates are different around the world.[2] Porphyria cutanea tarda is believed to be the most common type.[1] The disease was described as early as 370 BC by Hippocrates.[5] The underlying mechanism was first described by German physiologist and chemist Felix Hoppe-Seyler in 1871.[5] The name porphyria is from the Greek πορφύρα, porphyra, meaning "purple", a reference to the color of the urine that may be present during an attack.[5]
Signs and symptoms
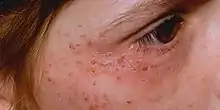
Acute porphyrias
Acute intermittent porphyria (AIP), variegate porphyria (VP), aminolevulinic acid dehydratase deficiency porphyria (ALAD) and hereditary coproporphyria (HCP). These diseases primarily affect the nervous system, resulting in episodic crises known as acute attacks. The major symptom of an acute attack is abdominal pain, often accompanied by vomiting, hypertension (elevated blood pressure), and tachycardia (an abnormally rapid heart rate).[4]
The most severe episodes may involve neurological complications: typically motor neuropathy (severe dysfunction of the peripheral nerves that innervate muscle), which leads to muscle weakness and potentially to quadriplegia (paralysis of all four limbs) and central nervous system symptoms such as seizures and coma. Occasionally, there may be short-lived psychiatric symptoms such as anxiety, confusion, hallucinations, and, very rarely, overt psychosis. All these symptoms resolve once the acute attack passes.
Given the many presentations and the relatively low occurrence of porphyria, patients may initially be suspected to have other, unrelated conditions. For instance, the polyneuropathy of acute porphyria may be mistaken for Guillain–Barré syndrome, and porphyria testing is commonly recommended in those situations.[6] Elevation of aminolevulinic acid from lead-induced disruption of heme synthesis results in lead poisoning having symptoms similar to acute porphyria.[7][8][9][10][11][12]
Chronic porphyrias
The non-acute porphyrias are X-linked dominant protoporphyria (XLDPP), congenital erythropoietic porphyria (CEP), porphyria cutanea tarda (PCT), and erythropoietic protoporphyria (EPP). None of these are associated with acute attacks; their primary manifestation is with skin disease. For this reason, these four porphyrias—along with two acute porphyrias, VP and HCP, that may also involve skin manifestations—are sometimes called cutaneous porphyrias.
Skin disease is encountered where excess porphyrins accumulate in the skin. Porphyrins are photoactive molecules, and exposure to light results in promotion of electrons to higher energy levels. When these return to the resting energy level or ground state, energy is released. This accounts for the property of fluorescence typical of the porphyrins. This causes local skin damage.
Two distinct patterns of skin disease are seen in porphyria:
- Immediate photosensitivity. This is typical of XLDPP and EPP. Following a variable period of sun exposure—typically about 30 minutes—patients complain of severe pain, burning, and discomfort in exposed areas. Typically, the effects are not visible, though occasionally there may be some redness and swelling of the skin.
- Vesiculo-erosive skin disease. This—a reference to the characteristic blistering (vesicles) and open sores (erosions) noted in patients—is the pattern seen in CEP, PCT, VP, and HCP. The changes are noted only in sun-exposed areas such as the face and back of the hands. Milder skin disease, such as that seen in VP and HCP, consists of increased skin fragility in exposed areas with a tendency to form blisters and erosions, particularly after minor knocks or scrapes. These heal slowly, often leaving small scars that may be lighter or darker than normal skin. More severe skin disease is sometimes seen in PCT, with prominent lesions, darkening of exposed skin such as the face, and hypertrichosis: abnormal hair growth on the face, particularly the cheeks. The most severe disease is seen in CEP and a rare variant of PCT known as hepatoerythropoietic porphyria (HEP); symptoms include severe shortening of digits, loss of skin appendages such as hair and nails, and severe scarring of the skin with progressive disappearance of ears, lips, and nose. Patients may also show deformed, discolored teeth or gum and eye abnormalities.
Cause
The porphyrias are generally considered genetic in nature.
Genetics
Subtypes of porphyrias depend on which enzyme is deficient.
| Porphyria type | Deficient enzyme | Type of porphyria | Inheritance | Symptoms | Prevalence |
|---|---|---|---|---|---|
| Aminolevulinate dehydratase deficiency porphyria (ALADP) | 5-aminolevulinate dehydratase (ALAD) | Hepatic | Autosomal recessive[13] | Abdominal pain, neuropathy[13] | Extremely rare; fewer than 10 cases ever reported.[14] |
| Acute intermittent porphyria (AIP) | Hydroxymethylbilane synthase (HMBS) formerly porphobilinogen deaminase (PBGD) | Hepatic | Autosomal dominant[13] | Periodic abdominal pain, peripheral neuropathy, psychiatric disorders, tachycardia[13] | 1 in 10,000[15]–20,000[15] |
| Congenital erythropoietic porphyria (CEP) | uroporphyrinogen synthase (UROS) | Erythropoietic | Autosomal recessive[13] | Severe photosensitivity with erythema, swelling and blistering. Hemolytic anemia, splenomegaly[13] | 1 in 1,000,000 or less.[16] |
| Porphyria cutanea tarda (PCT) | uroporphyrinogen decarboxylase (UROD) | Hepatic | Approximately 80% sporadic,[17] 20% Autosomal dominant[13] | Photosensitivity with vesicles and bullae[13] | 1 in 10,000[18] |
| Hereditary coproporphyria (HCP) | coproporphyrinogen oxidase (CPOX) | Hepatic | Autosomal dominant[13] | Photosensitivity, neurologic symptoms, colic[13] | 1 in 500,000[18] |
| Harderoporphyria | coproporphyrinogen oxidase (CPOX) | Erythropoietic | Autosomal recessive[13] | Jaundice, anemia, enlarged liver and spleen, often neonatal. Photosensitivity later. | Extremely rare; fewer than 10 cases ever reported. |
| Variegate porphyria (VP) | protoporphyrinogen oxidase (PPOX) | Hepatic | Autosomal dominant[19] | Photosensitivity, neurologic symptoms, developmental delay | 1 in 300 in South Africa[18] 1 in 75,000 in Finland[20] |
| Erythropoietic protoporphyria (EPP) | ferrochelatase (FECH) | Erythropoietic | Autosomal recessive[13] | Photosensitivity with skin lesions. Gallstones, mild liver dysfunction[13] | 1 in 75,000[18]–200,000[18] |
X-linked dominant protoporphyria is a rare form of erythropoietic protoporphyria caused by a gain-of-function mutation in ALAS2 characterized by severe photosensitivity.[21][22]
In the autosomal recessive types, if a person inherits a single gene they may become a carrier. Generally they do not have symptoms, but may pass the gene onto offspring.[23]
Triggers
Acute porphyria can be triggered by a number of drugs, most of which are believed to trigger it by interacting with enzymes in the liver which are made with heme. Such drugs include:[24][25][26]
- Sulfonamides, including sulfadiazine, sulfasalazine and trimethoprim/sulfamethoxazole.
- Sulfonylureas like glibenclamide, gliclazide and glimepiride, although glipizide is thought to be safe.
- Barbiturates including thiopental, phenobarbital, primidone, etc.
- Systemic treatment with antifungals including fluconazole, griseofulvin, ketoconazole and voriconazole. (Topical use of these agents is thought to be safe due to minimal systemic absorption.)
- Certain antibiotics like rifapentine, rifampicin, rifabutine, isoniazid, nitrofurantoin and, possibly, metronidazole.
- Ergot derivatives including dihydroergotamine, ergometrine, ergotamine, methysergide, etc.
- Certain antiretroviral medications (e.g. indinavir, nevirapine, ritonavir, saquinavir, etc.)
- Progestogens
- Some anticonvulsants including: carbamazepine, ethosuximide, phenytoin, topiramate, valproate.
- Some painkillers like dextropropoxyphene, ketorolac, metamizole, pentazocine
- Some cancer treatments like bexarotene, busulfan, chlorambucil, estramustine, etoposide, flutamide, idarubicin, ifosfamide, irinotecan, ixabepilone, letrozole, lomustine, megestrol, mitomycin, mitoxantrone, paclitaxel, procarbazine, tamoxifen, topotecan
- Some antidepressants like imipramine, phenelzine, trazodone
- Some antipsychotics like risperidone, ziprasidone
- Some retinoids used for skin conditions like acitretin and isotretinoin
- Miscellaneous others including: cocaine, methyldopa, fenfluramine, disulfiram, orphenadrine, pentoxifylline, and sodium aurothiomalate.
Pathogenesis
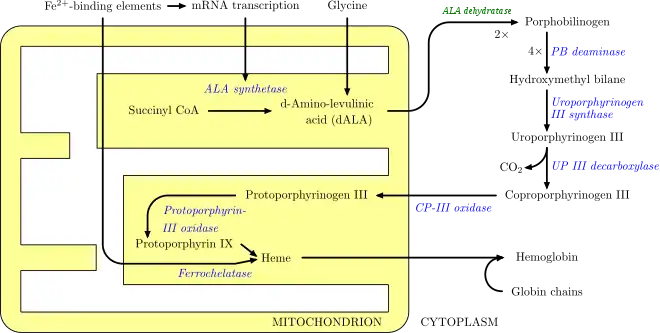
In humans, porphyrins are the main precursors of heme, an essential constituent of hemoglobin, myoglobin, catalase, peroxidase, and P450 liver cytochromes.[27]
The body requires porphyrins to produce heme, which is used to carry oxygen in the blood among other things, but in the porphyrias there is a deficiency (inherited or acquired) of the enzymes that transform the various porphyrins into others, leading to abnormally high levels of one or more of these substances. Porphyrias are classified in two ways, by symptoms and by pathophysiology. Physiologically, porphyrias are classified as liver or erythropoietic based on the sites of accumulation of heme precursors, either in the liver or in the bone marrow and red blood cells.[28]
Deficiency in the enzymes of the porphyrin pathway leads to insufficient production of heme. Heme function plays a central role in cellular metabolism. This is not the main problem in the porphyrias; most heme synthesis enzymes—even dysfunctional enzymes—have enough residual activity to assist in heme biosynthesis. The principal problem in these deficiencies is the accumulation of porphyrins, the heme precursors, which are toxic to tissue in high concentrations. The chemical properties of these intermediates determine the location of accumulation, whether they induce photosensitivity, and whether the intermediate is excreted (in the urine or feces).
There are eight enzymes in the heme biosynthetic pathway, four of which—the first one and the last three—are in the mitochondria, while the other four are in the cytosol. Defects in any of these can lead to some form of porphyria. The hepatic porphyrias are characterized by acute neurological attacks (seizures, psychosis, extreme back and abdominal pain, and an acute polyneuropathy), while the erythropoietic forms present with skin problems, usually a light-sensitive blistering rash and increased hair growth. Variegate porphyria (also porphyria variegata or mixed porphyria), which results from a partial deficiency in PROTO oxidase, manifests itself with skin lesions similar to those of porphyria cutanea tarda combined with acute neurologic attacks. Hereditary coproporphyria, which is characterized by a deficiency in coproporphyrinogen oxidase, coded for by the CPOX gene, may also present with both acute neurologic attacks and cutaneous lesions. All other porphyrias are either skin- or nerve-predominant.[29]
Diagnosis
Porphyrin studies
Porphyria is diagnosed through biochemical analysis of blood, urine, and stool.[16][30] In general, urine estimation of porphobilinogen (PBG) is the first step if acute porphyria is suspected. As a result of feedback, the decreased production of heme leads to increased production of precursors, PBG being one of the first substances in the porphyrin synthesis pathway.[31] In nearly all cases of acute porphyria syndromes, urinary PBG is markedly elevated except for the very rare ALA dehydratase deficiency or in patients with symptoms due to hereditary tyrosinemia type I.[32] In cases of mercury- or arsenic poisoning-induced porphyria, other changes in porphyrin profiles appear, most notably elevations of uroporphyrins I & III, coproporphyrins I & III, and pre-coproporphyrin.[33]
Repeat testing during an attack and subsequent attacks may be necessary in order to detect a porphyria, as levels may be normal or near-normal between attacks. The urine screening test has been known to fail in the initial stages of a severe, life-threatening attack of acute intermittent porphyria.
Up to 90% of the genetic carriers of the more common, dominantly inherited acute hepatic porphyrias (acute intermittent porphyria, hereditary coproporphyria, variegate porphyria) have been noted in DNA tests to be latent for classic symptoms and may require DNA or enzyme testing. The exception to this may be latent post-puberty genetic carriers of hereditary coproporphyria.
As most porphyrias are rare conditions, general hospital labs typically do not have the expertise, technology, or staff time to perform porphyria testing. In general, testing involves sending samples of blood, stool, and urine to a reference laboratory.[16] All samples to detect porphyrins must be handled properly. Samples should be taken during an acute attack; otherwise a false negative result may occur. Samples must be protected from light and either refrigerated or preserved.[16]
If all the porphyrin studies are negative, one must consider pseudoporphyria. A careful medication review often will find the cause of pseudoporphyria.
Additional tests
Further diagnostic tests of affected organs may be required, such as nerve conduction studies for neuropathy or an ultrasound of the liver. Basic biochemical tests may assist in identifying liver disease, hepatocellular carcinoma, and other organ problems.[34]
Management
Acute porphyria
Carbohydrate administration
Often, empirical treatment is required if the diagnostic suspicion of a porphyria is high since acute attacks can be fatal. A high-carbohydrate diet is typically recommended; in severe attacks, a dextrose 10% infusion is commenced, which may aid in recovery by suppressing heme synthesis, which in turn reduces the rate of porphyrin accumulation. However, this can worsen cases of low blood sodium levels (hyponatraemia) and should be done with extreme caution as it can prove fatal.[35]
Heme analogs
Hematin (trade name Panhematin) and heme arginate (trade name NormoSang) are the drugs of choice in acute porphyria, in the United States and the United Kingdom, respectively. These drugs need to be given very early in an attack to be effective; effectiveness varies amongst individuals. They are not curative drugs but can shorten attacks and reduce the intensity of an attack. Side effects are rare but can be serious. These heme-like substances theoretically inhibit ALA synthase and hence the accumulation of toxic precursors. In the United Kingdom, supplies of NormoSang are kept at two national centers; emergency supply is available from St Thomas's Hospital, London.[36] In the United States, Lundbeck manufactures and supplies Panhematin for infusion.[37]
Heme arginate (NormoSang) is used during crises but also in preventive treatment to avoid crises, one treatment every 10 days.
Any sign of low blood sodium (hyponatremia) or weakness should be treated with the addition of hematin, heme arginate, or even tin mesoporphyrin, as these are signs of impending syndrome of inappropriate antidiuretic hormone (SIADH) or peripheral nervous system involvement that may be localized or severe, progressing to bulbar paresis and respiratory paralysis.
Cimetidine
Cimetidine has also been reported to be effective for acute porphyric crisis and possibly effective for long-term prophylaxis.[38]
Symptom control
Pain is severe, frequently out of proportion to physical signs, and often requires the use of opiates to reduce it to tolerable levels. Pain should be treated as early as medically possible. Nausea can be severe; it may respond to phenothiazine drugs but is sometimes intractable. Hot baths and showers may lessen nausea temporarily, though caution should be used to avoid burns or falls.
Early identification
It is recommended that patients with a history of acute porphyria, and even genetic carriers, wear an alert bracelet or other identification at all times. This is in case they develop severe symptoms, or in case of accidents where there is a potential for drug exposure, and as a result they are unable to explain their condition to healthcare professionals. Some drugs are absolutely contraindicated for patients with any form of porphyria.[39]
Neurologic and psychiatric disorders
Patients who experience frequent attacks can develop chronic neuropathic pain in extremities as well as chronic pain in the abdomen.[40] Intestinal pseudo-obstruction, ileus, intussusception, hypoganglionosis, and encopresis in children have been associated with porphyrias. This is thought to be due to axonal nerve deterioration in affected areas of the nervous system and vagal nerve dysfunction. Pain treatment with long-acting opioids, such as morphine, is often indicated, and, in cases where seizure or neuropathy is present, gabapentin is known to improve outcome.[41]
Seizures often accompany this disease. Most seizure medications exacerbate this condition. Treatment can be problematic: barbiturates especially must be avoided. Some benzodiazepines are safe and, when used in conjunction with newer anti-seizure medications such as gabapentin, offer a possible regimen for seizure control. Gabapentin has the additional feature of aiding in the treatment of some kinds of neuropathic pain.[41] Magnesium sulfate and bromides have also been used in porphyria seizures; however, development of status epilepticus in porphyria may not respond to magnesium alone. The addition of hematin or heme arginate has been used during status epilepticus.[42]
Depression often accompanies the disease and is best dealt with by treating the offending symptoms and if needed the judicious use of antidepressants. Some psychotropic drugs are porphyrinogenic, limiting the therapeutic scope. Other psychiatric symptoms such as anxiety, restlessness, insomnia, depression, mania, hallucinations, delusions, confusion, catatonia, and psychosis may occur.[43]
Underlying liver disease
Some liver diseases may cause porphyria even in the absence of genetic predisposition. These include hemochromatosis and hepatitis C. Treatment of iron overload may be required.[2]
Patients with the acute porphyrias (AIP, HCP, VP) are at increased risk over their life for hepatocellular carcinoma (primary liver cancer) and may require monitoring. Other typical risk factors for liver cancer need not be present.[2]
Hormone treatment
Hormonal fluctuations that contribute to cyclical attacks in women have been treated with oral contraceptives and luteinizing hormones to shut down menstrual cycles. However, oral contraceptives have also triggered photosensitivity and withdrawal of oral contraceptives has triggered attacks. Androgens and fertility hormones have also triggered attacks.[44] In 2019, givosiran was approved in the United States for the treatment of acute hepatic porphyria.[45][46]
Erythropoietic porphyria
These are associated with accumulation of porphyrins in erythrocytes and are rare.
The pain, burning, swelling, and itching that occur in erythropoietic porphyrias generally require avoidance of bright sunlight. Most kinds of sunscreen are not effective, but SPF-rated long-sleeve shirts, hats, bandanas, and gloves can help. Chloroquine may be used to increase porphyrin secretion in some EPs.[16] Blood transfusion is occasionally used to suppress innate heme production.
The rarest is congenital erythropoietic porphyria (CEP), otherwise known as Gunther's disease. The signs may present from birth and include severe photosensitivity, brown teeth that fluoresce in ultraviolet light due to deposition of Type 1 porphyrins, and later hypertrichosis. Hemolytic anemia usually develops. Pharmaceutical-grade beta carotene may be used in its treatment.[47] A bone marrow transplant has also been successful in curing CEP in a few cases, although long-term results are not yet available.[48]
In December 2014, afamelanotide received authorization from the European Commission as a treatment for the prevention of phototoxicity in adult patients with EPP.[49] In a 2023 industry-funded phase 2 trial, dersimelagon, an orally administered, selective melanocortin 1 receptor agonist that increases levels of skin eumelanin, was reported to have increased the duration of symptom-free sunlight exposure and quality of life compared to placebo in patients with erythropoietic protoporphyria.[50]
Epidemiology
Rates of all types of porphyria taken together have been estimated to be approximately one in 25,000 in the United States.[51] The worldwide prevalence has been estimated to be between one in 500 and one in 50,000 people.[52]
Porphyrias have been detected in all races and in multiple ethnic groups on every continent. There are high incidence reports of AIP in areas of India and Scandinavia. More than 200 genetic variants of AIP are known, some of which are specific to families, although some strains have proven to be repeated mutations.
History
The underlying mechanism was first described by the German physiologist Felix Hoppe-Seyler in 1871,[53] and acute porphyrias were described by the Dutch physician Barend Stokvis in 1889.[54][55]
The links between porphyrias and mental illness have been noted for decades. In the early 1950s, patients with porphyrias (occasionally referred to as "porphyric hemophilia"[56]) and severe symptoms of depression or catatonia were treated with electroshock therapy.
Vampires and werewolves
Porphyria has been suggested as an explanation for the origin of vampire and werewolf legends, based upon certain perceived similarities between the condition and the folklore.
In January 1964, L. Illis's 1963 paper, "On Porphyria and the Aetiology of Werewolves," was published in Proceedings of the Royal Society of Medicine. Later, Nancy Garden argued for a connection between porphyria and the vampire belief in her 1973 book, Vampires. In 1985, biochemist David Dolphin's paper for the American Association for the Advancement of Science, "Porphyria, Vampires, and Werewolves: The Aetiology of European Metamorphosis Legends," gained widespread media coverage, popularizing the idea.
The theory has been rejected by a few folklorists and researchers as not accurately describing the characteristics of the original werewolf and vampire legends or the disease, and as potentially stigmatizing people with porphyria.[57][58]
A 1995 article from the Postgraduate Medical Journal (via NIH) explains:
As it was believed that the folkloric vampire could move about freely in daylight hours, as opposed to the 20th century variant, congenital erythropoietic porphyria cannot readily explain the folkloric vampire but may be an explanation of the vampire as we know it in the 20th century. In addition, the folkloric vampire, when unearthed, was always described as looking quite healthy ("as they were in life"), while due to disfiguring aspects of the disease, sufferers would not have passed the exhumation test. Individuals with congenital erythropoietic porphyria do not crave blood. The enzyme (hematin) necessary to alleviate symptoms is not absorbed intact on oral ingestion, and drinking blood would have no beneficial effect on the sufferer. Finally, and most important, the fact that vampire reports were literally rampant in the 18th century, and that congenital erythropoietic porphyria is an extremely rare manifestation of a rare disease, makes it an unlikely explanation of the folkloric vampire.[59]
Notable cases
- George III. The mental illness exhibited by George III in the regency crisis of 1788 has inspired several attempts at retrospective diagnosis. The first, written in 1855, thirty-five years after his death, concluded that he had acute mania. M. Guttmacher, in 1941, suggested manic-depressive psychosis as a more likely diagnosis. The first suggestion that a physical illness was the cause of King George's mental derangement came in 1966, in a paper called "The Insanity of King George III: A Classic Case of Porphyria",[60] with a follow-up in 1968, "Porphyria in the Royal Houses of Stuart, Hanover and Prussia".[61] The papers, by a mother/son psychiatrist team, were written as though the case for porphyria had been proven, but the response demonstrated that many experts, including those more intimately familiar with the manifestations of porphyria, were unconvinced. Many psychiatrists disagreed with the diagnosis, suggesting bipolar disorder as far more probable. The theory is treated in Purple Secret,[62] which documents the ultimately unsuccessful search for genetic evidence of porphyria in the remains of royals suspected to have had it.[63] In 2005, it was suggested that arsenic (which is known to be porphyrogenic) given to George III with antimony may have caused his porphyria.[64] This study found high levels of arsenic in King George's hair. In 2010, one analysis of historical records argued that the porphyria claim was based on spurious and selective interpretation of contemporary medical and historical sources.[65] The mental illness of George III is the basis of the plot in The Madness of King George, a 1994 British film based upon the 1991 Alan Bennett play, The Madness of George III. The closing credits of the film include the comment that the King's symptoms suggest that he had porphyria, and note that the disease is "periodic, unpredictable, and hereditary". The traditional argument that George III did not have porphyria, but rather bipolar disorder, is thoroughly defended by Andrew Roberts in his new biography The Last King of America.[66]
- Descendants of George III. Among other descendants of George III theorized by the authors of Purple Secret to have had porphyria (based on analysis of their extensive and detailed medical correspondence) were his great-great-granddaughter Princess Charlotte of Prussia (Emperor William II's eldest sister) and her daughter Princess Feodora of Saxe-Meiningen. They uncovered better evidence that George III's great-great-great-grandson Prince William of Gloucester was reliably diagnosed with variegate porphyria.[67]
- Mary, Queen of Scots. It is believed that Mary, Queen of Scots, King George III's ancestor, also had acute intermittent porphyria,[68] although this is subject to much debate. It is assumed she inherited the disorder, if indeed she had it, from her father, James V of Scotland. Both father and daughter endured well-documented attacks that could fall within the constellation of symptoms of porphyria.
- Maria I of Portugal. Maria I—known as "Maria the Pious" or "Maria the Mad" because of both her religious fervor and her acute mental illness, which made her incapable of handling state affairs after 1792—is also thought to have had porphyria. Francis Willis, the same physician who treated George III, was even summoned by the Portuguese court but returned to England after the court limited the treatments he could oversee. Contemporary sources, such as Secretary of State for Foreign Affairs Luís Pinto de Sousa Coutinho, noted that the queen had ever-worsening stomach pains and abdominal spasms: hallmarks of porphyria.[69]
- Vlad III Dracula, "The Impaler." Vlad III was also said to have had acute porphyria, which may have started the notion that vampires were allergic to sunlight.[70]
- Vincent van Gogh. Other commentators have suggested that Vincent van Gogh may have had acute intermittent porphyria.[71]
- King Nebuchadnezzar of Babylon. The description of this king in Daniel 4 suggests to some that he had porphyria.
- Physician Archie Cochrane. He was born with porphyria, which caused health problems throughout his life.[72]
- Paula Frías Allende. The daughter of the Chilean novelist Isabel Allende. She fell into a porphyria-induced coma in 1991,[73] which inspired Isabel to write the memoir Paula, dedicated to her.
 George III in his coronation robes
George III in his coronation robes Mary, Queen of Scots c. 1578.
Mary, Queen of Scots c. 1578._-_Google_Cultural_Institute.jpg.webp)
Uses in literature
Stated or implied references to porphyria are included in some literature, particularly gothic literature. These include the following:
- The condition is the name of the title character in the gothic poem "Porphyria's Lover," by Robert Browning.
- The condition is heavily implied to be the cause of the symptoms suffered by the narrator in the gothic short story "Lusus Naturae," by Margaret Atwood. Some of the narrator's symptoms resemble those of porphyria, and one passage of the story states that the name of the narrator's disease "had some Ps and Rs in it."
References
- 1 2 3 4 5 6 7 8 9 10 11 "porphyria". MedlinePlus. July 2009. Retrieved 31 March 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 "Porphyria | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Retrieved 6 December 2018.
- ↑ Dancygier, Henryk (2009). Clinical Hepatology: Principles and Practice of Hepatobiliary Diseases. Springer Science & Business Media. p. 1088. ISBN 9783642045196. Archived from the original on 8 September 2017.
- 1 2 3 4 Stein, PE; Badminton, MN; Rees, DC (February 2017). "Update review of the acute porphyrias". British Journal of Haematology. 176 (4): 527–538. doi:10.1111/bjh.14459. PMID 27982422. S2CID 19970176.
- 1 2 3 McManus, Linda (2014). Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms. Elsevier. p. 1488. ISBN 9780123864574. Archived from the original on 8 September 2017.
- ↑ Albers JW, Fink JK (2004). "Porphyric neuropathy" (PDF). Muscle Nerve. 30 (4): 410–422. doi:10.1002/mus.20137. hdl:2027.42/34640. PMID 15372536. S2CID 68067335.
- ↑ Vannotti A (1954). Porphyrins: Their Biological and Chemical Importance. Hilger & Watts, Hilger Division. p. 126.
Indeed, lead poisoning, like all porphyrin diseases, is accompanied by obstinate constipation, nervous lesions, hyperpigmentation and abdominal attacks.
- ↑ Dancygier H (2009). Clinical Hepatology: Principles and Practice of Hepatobiliary Diseases. Springer Science & Business Media. p. 1088. ISBN 9783642045196. Archived from the original on 8 September 2017.
- ↑ Akshatha LN, Rukmini MS, Mamatha TS, Sadashiva Rao P, Prashanth B (December 2014). "Lead poisoning mimicking acute porphyria!". Journal of Clinical and Diagnostic Research. 8 (12): CD01-2. doi:10.7860/JCDR/2014/10597.5315. PMC 4316248. PMID 25653942.
- ↑ Tsai MT, Huang SY, Cheng SY (2017). "Lead Poisoning Can Be Easily Misdiagnosed as Acute Porphyria and Nonspecific Abdominal Pain". Case Reports in Emergency Medicine. 2017: 9050713. doi:10.1155/2017/9050713. PMC 5467293. PMID 28630774.
- ↑ Wang, B.; Bissell, D. M.; Adam, M. P.; Ardinger, H. H.; Pagon, R. A.; Wallace, S. E.; Bean LJH; Stephens, K.; Amemiya, A. (2018). "Hereditary Coproporphyria". GeneReviews. PMID 23236641. Retrieved 28 February 2020.
the symptoms in lead poisoning closely mimic those of acute porphyria
- ↑ Murphy, Brian (3 November 2020). "What Tests Can Help Diagnose Porphyria?". Retrieved 22 May 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Table 18-1 in: Marks, Dawn B.; Swanson, Todd; Sandra I Kim; Marc Glucksman (2007). Biochemistry and molecular biology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 978-0-7817-8624-9.
- ↑ Overview of the Porphyrias Archived 22 July 2011 at the Wayback Machine at The Porphyrias Consortium (a part of NIH Rare Diseases Clinical Research Network (RDCRN)) Retrieved June 2011
- 1 2 Medscape - Diseases of Tetrapyrrole Metabolism - Refsum Disease and the Hepatic Porphyrias Archived 4 June 2011 at the Wayback Machine Author: Norman C Reynolds. Chief Editor: Stephen A Berman. Updated: 23 March 2009
- 1 2 3 4 5 Thadani H, Deacon A, Peters T (2000). "Diagnosis and management of porphyria". BMJ. 320 (7250): 1647–1651. doi:10.1136/bmj.320.7250.1647. PMC 1127427. PMID 10856069.
- ↑ "OMIM Entry - 176090 - PORPHYRIA CUTANEA TARDA, TYPE I". omim.org. Retrieved 24 February 2021.
- 1 2 3 4 5 Arceci, Robert.; Hann, Ian M.; Smith, Owen P. (2006). Pediatric hematolog. Malden, Mass.: Blackwell Pub. ISBN 978-1-4051-3400-2.
- ↑ Singal, AK; Anderson, KE; Pagon, RA; Adam, MP; Ardinger, HH; Wallace, SE; Amemiya, A; Bean, LJH; Bird, TD; Dolan, CR; Fong, CT; Smith, RJH; Stephens, K (2013). "Variegate Porphyria". GeneReviews. Seattle, USA: University of Washington. PMID 23409300. Archived from the original on 18 January 2017. Retrieved 18 April 2015.
- ↑ Mustajoki P (1980). "Variegate porphyria. Twelve years' experience in Finland". The Quarterly Journal of Medicine. 49 (194): 191–203. PMID 7433635.
- ↑ "OMIM Entry - # 300752 - PROTOPORPHYRIA, ERYTHROPOIETIC, X-LINKED; XLEPP". omim.org.
- ↑ "Orphanet: X linked erythropoietic protoporphyria". www.orpha.net.
- ↑ "Porphyria - Symptoms and causes". Mayo Clinic. Retrieved 5 March 2020.
- ↑ Tishler, PV. "About Porphyria: Safety database". Porphyria Drug Safety Database. American Porphyria Foundation. Archived from the original on 12 July 2017. Retrieved 27 August 2017.
- ↑ Brun, A. "napos". The Drug Database for Acute Porphyria. European Porphyria Network. Archived from the original on 23 August 2017. Retrieved 27 August 2017.
- ↑ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. p. 663. ISBN 978-0-85711-084-8.
- ↑ Kumari, Asha (12 October 2017). Sweet Biochemistry. Elsevier Science. ISBN 9780128144534. Retrieved 30 October 2022.
- ↑ Lourenço, Charles Marquez; Lee, Chul; Anderson, Karl E. (2012). "Disorders of Haem Biosynthesis". In Saudubray, Jean-Marie; van den Berghe, Georges; Walter, John H. (eds.). Inborn Metabolic Diseases: Diagnosis and Treatment (5th ed.). New York: Springer. pp. 521–532. ISBN 978-3-642-15719-6.
- ↑ Ogun, Aminat S.; Joy, Neena V.; Valentine, Menogh (2022), "Biochemistry, Heme Synthesis", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30726014, retrieved 30 October 2022
- ↑ "Tests for Porphyria diagnosis". American Porphyria Foundation. Archived from the original on 20 March 2014. Retrieved 17 May 2014.
- ↑ Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ (2005). "Recommendations for the diagnosis and treatment of the acute porphyrias". Ann. Intern. Med. 142 (6): 439–50. doi:10.7326/0003-4819-142-6-200503150-00010. PMID 15767622. S2CID 36122555.
- ↑ Berkó G, Durkó I (19 December 1971). "[Modification of the Mauzerall-Granick method for the determination of urinary aminolevulinic acid]". Orvosi Hetilap (in Hungarian). 112 (51): 3085–6. PMID 5136653.
- ↑ Woods, J.S. (1995). "Porphyrin metabolism as indicator of metal exposure and toxicity". In Goyer, R.A. & Cherian, M.G. (ed.). Toxicology of metals, biochemical aspects. Vol. 115. Berlin: Springer. pp. 19–52, Chapter 2.
{{cite book}}: CS1 maint: multiple names: editors list (link) - ↑ Di Pierro, Elena; De Canio, Michele; Mercadante, Rosa; Savino, Maria; Granata, Francesca; Tavazzi, Dario; Nicolli, Anna Maria; Trevisan, Andrea; Marchini, Stefano; Fustinoni, Silvia (26 July 2021). "Laboratory Diagnosis of Porphyria". Diagnostics. 11 (8): 1343. doi:10.3390/diagnostics11081343. ISSN 2075-4418. PMC 8391404. PMID 34441276.
- ↑ patient.co.uk. "Porphyrias - immediate management". Archived from the original on 20 October 2016. Retrieved 19 October 2016.
- ↑ British Medical Association, Royal Pharmaceutical Society of Great Britain (March 2009). "9.8.2: Acute porphyrias". British National Formulary (BNF 57). United Kingdom: BMJ Group and RPS Publishing. p. 549. ISBN 978-0-85369-845-6.
- ↑ American Porphyria Foundation (2010). "Panhematin for Acute Porphyria". Archived from the original on 25 August 2010. Retrieved 5 August 2010.
- ↑ Cherem JH, Malagon J, Nellen H (2005). "Cimetidine and acute intermittent porphyria". Ann. Intern. Med. 143 (9): 694–5. doi:10.7326/0003-4819-143-9-200511010-00023. PMID 16263899.
- ↑ "Porphyria - Diagnosis and treatment - Mayo Clinic". www.mayoclinic.org. Retrieved 6 December 2018.
- ↑ Birgisdottir BT, Asgeirsson H, Arnardottir S, Jonsson JJ, Vidarsson B (2010). "[Acute abdominal pain caused by acute intermittent porphyria - case report and review of the literature]". Laeknabladid (in Icelandic). 96 (6): 413–18. PMID 20519771.
- 1 2 Tsao YC, Niu DM, Chen JT, Lin CY, Lin YY, Liao KK (2010). "Gabapentin reduces neurovisceral pain of porphyria". Acta Neurol Taiwan. 19 (2): 112–5. PMID 20714961.
- ↑ Cherian A, Thomas SV (2009). "Status epilepticus". Ann Indian Acad Neurol. 12 (3): 140–53. doi:10.4103/0972-2327.56312. PMC 2824929. PMID 20174493.
- ↑ Murray ED, Buttner N, Price BH. (2012) Depression and Psychosis in Neurological Practice. In: Neurology in Clinical Practice, 6th Edition. Bradley WG, Daroff RB, Fenichel GM, Jankovic J (eds.) Butterworth Heinemann. 12 April 2012. ISBN 978-1437704341
- ↑ "Hormonal Contraceptives and Porphyria". Porphyria news. September 2020.
- ↑ "FDA approves first treatment for inherited rare disease". U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ↑ "FDA approves givosiran for acute hepatic porphyria". U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019. This article incorporates text from this source, which is in the public domain.
- ↑ Martin A Crook.2006. Clinical chemistry and Metabolic Medicine. seventh edition. Hodder Arnold. ISBN 0-340-90616-2
- ↑ Faraci M, Morreale G, Boeri E, Lanino E, Dallorso S, Dini G, Scuderi F, Cohen A, Cappelli B (2008). "Unrelated HSCT in an adolescent affected by congenital erythropoietic porphyria". Pediatr Transplant. 12 (1): 117–120. doi:10.1111/j.1399-3046.2007.00842.x. PMID 18186900. S2CID 20789520.
- ↑ "Community register of medicinal products for human use". ec.europa.eu. European Commission. Archived from the original on 24 December 2014. Retrieved 24 December 2014.
- ↑ Balwani, Manisha; Bonkovsky, Herbert L.; Levy, Cynthia; Anderson, Karl E.; Bissell, D. Montgomery; Parker, Charles; Takahashi, Fumihiro; Desnick, Robert J.; Belongie, Kirstine (13 April 2023). "Dersimelagon in Erythropoietic Protoporphyrias". New England Journal of Medicine. 388 (15): 1376–1385. doi:10.1056/NEJMoa2208754. ISSN 0028-4793. PMID 37043653. S2CID 258110676.
- ↑ eMedicine > Porphyria, Cutaneous Archived 5 December 2010 at the Wayback Machine Authors: Vikramjit S Kanwar, Thomas G DeLoughery, Richard E Frye and Darius J Adams. Updated: 27 July 2010.
- ↑ Genetics Home Reference > Porphyria Archived 5 August 2010 at the Wayback Machine Reviewed July 2009
- ↑ Hoppe-Seyler F (1871). "Das Hämatin". Tubinger Med-Chem Untersuch. 4: 523–33.
- ↑ Nick Lane (16 December 2002). "Born to the purple: the story of porphyria". Scientific American. Archived from the original on 11 October 2007. Retrieved 5 August 2008.
- ↑ Stokvis BJ. "Over twee zeldzame kleurstoffen in urine van zieken". Nederl Tijdschr Geneeskd (in Dutch). 2: 409–417. Reprinted in Stokvis BJ (December 1989). "Over twee zeldzame kleurstoffen in urine van zieken". Ned Tijdschr Geneeskd (in Dutch). 133 (51): 2562–70. PMID 2689889.
- ↑ Denver, Joness. "An Encyclopaedia of Obscure Medicine". Published by University Books, Inc., 1959.
- ↑ American Vampires: Fans, Victims, Practitioners, Norine Dresser, W. W. Norton & Company, 1989.
- ↑ "Did vampires suffer from the disease porphyria — or not?" Archived 5 February 2013 at the Wayback Machine The Straight Dope, 7 May 1999.
- ↑ Cox, Ann M. (1995). "Porphyria and vampirism: another myth in the making". Postgraduate Medical Journal. 71 (841): 643–644. doi:10.1136/pgmj.71.841.643-a. PMC 2398345. PMID 7494765.
- ↑ Macalpine I, Hunter R (January 1966). "The 'insanity' of King George 3d: a classic case of porphyria". Br Med J. 1 (5479): 65–71. doi:10.1136/bmj.1.5479.65. PMC 1843211. PMID 5323262.
- ↑ Macalpine I, Hunter R, Rimington C (January 1968). "Porphyria in the royal houses of Stuart, Hanover, and Prussia. A follow-up study of George 3d's illness". Br Med J. 1 (5583): 7–18. doi:10.1136/bmj.1.5583.7. PMC 1984936. PMID 4866084.
- ↑ Warren, Martin; Rh̲l, John C. G.; Hunt, David C. (1998). Purple Secret: Genes, "Madness" and the Royal Houses of Europe. London: Bantam Books. ISBN 978-0-593-04148-2.
- ↑ The authors demonstrated a single point mutation in the PPOX gene but not one that has been associated with disease.
- ↑ Cox TM, Jack N, Lofthouse S, Watling J, Haines J, Warren MJ (2005). "King George III and porphyria: an elemental hypothesis and investigation". The Lancet. 366 (9482): 332–335. doi:10.1016/S0140-6736(05)66991-7. PMID 16039338. S2CID 13109527.
- ↑ Peters TJ, Wilkinson D (2010). "King George III and porphyria: a clinical re-examination of the historical evidence". History of Psychiatry. 21 (81 Pt 1): 3–19. doi:10.1177/0957154X09102616. PMID 21877427. S2CID 22391207.
- ↑ Roberts, Andrew (9 November 2021). "The Last King of America: The Misunderstood Reign of George III". Viking – via Amazon.
- ↑ Smith, Martin; Smith, Alison (21 December 2009). Tetrapyrroles: Birth, Life and Death. Springer. ISBN 9780387785189. Archived from the original on 4 July 2014. Retrieved 9 April 2014.
- ↑ Röhl, John (25 June 1999). "The Royal Family's Toxic Time-Bomb". Bulletin: The University of Sussex Newsletter. Archived from the original on 15 September 2013. Retrieved 18 August 2013.
- ↑ Roberts, Jenifer (2009). The Madness of Queen Maria [The Remarkable Life of Maria I of Portugal]. Templeton Press. ISBN 978-0954558918.
- ↑ "BBC News - UK - Charles visits 'relative' Dracula's home". Archived from the original on 24 August 2013.
- ↑ Loftus LS, Arnold WN (1991). "Vincent van Gogh's illness: acute intermittent porphyria?". BMJ. 303 (6817): 1589–1591. doi:10.1136/bmj.303.6817.1589. PMC 1676250. PMID 1773180.
- ↑ Cochrane, Archibald L; Blythe, Max (2009) [1989]. One Man's Medicine: An Autobiography of Professor Archie Cochrane. Cardiff: Cardiff University. ISBN 978-0-9540884-3-9.
- ↑ Allende, Isabel (1995). Paula. New York: HarperCollins. ISBN 978-0-06-017253-4.