In mass spectrometry, de novo peptide sequencing is the method in which a peptide amino acid sequence is determined from tandem mass spectrometry.
Knowing the amino acid sequence of peptides from a protein digest is essential for studying the biological function of the protein. In the old days, this was accomplished by the Edman degradation procedure.[1] Today, analysis by a tandem mass spectrometer is a more common method to solve the sequencing of peptides. Generally, there are two approaches: database search and de novo sequencing. Database search is a simple version as the mass spectra data of the unknown peptide is submitted and run to find a match with a known peptide sequence, the peptide with the highest matching score will be selected.[2] This approach fails to recognize novel peptides since it can only match to existing sequences in the database. De novo sequencing is an assignment of fragment ions from a mass spectrum. Different algorithms[3] are used for interpretation and most instruments come with de novo sequencing programs.
Peptide fragmentation
Peptides are protonated in positive-ion mode. The proton initially locates at the N-terminus or a basic residue side chain, but because of the internal solvation, it can move along the backbone breaking at different sites which result in different fragments. The fragmentation rules are well explained by some publications.[4][5][6][7][8][9]
Three different types of backbone bonds can be broken to form peptide fragments: alkyl carbonyl (CHR-CO), peptide amide bond (CO-NH), and amino alkyl bond (NH-CHR).
Different types of fragment ions
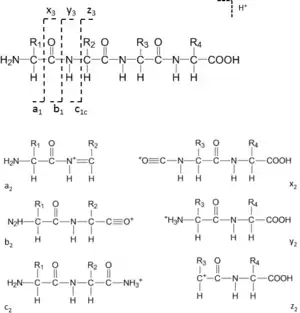
When the backbone bonds cleave, six different types of sequence ions are formed as shown in Fig. 1. The N-terminal charged fragment ions are classed as a, b or c, while the C-terminal charged ones are classed as x, y or z. The subscript n is the number of amino acid residues. The nomenclature was first proposed by Roepstorff and Fohlman, then Biemann modified it and this became the most widely accepted version.[11][12]
Among these sequence ions, a, b and y-ions are the most common ion types, especially in the low-energy collision-induced dissociation (CID) mass spectrometers, since the peptide amide bond (CO-NH) is the most vulnerable and the loss of CO from b-ions.
Mass of b-ions = Σ (residue masses) + 1 (H+)
Mass of y-ions = Σ (residue masses) + 19 (H2O+H+)
Mass of a-ions = mass of b-ions – 28 (CO)
Double backbone cleavage produces internal ions, acylium-type like H2N-CHR2-CO-NH-CHR3-CO+ or immonium-type like H2N-CHR2-CO-NH+=CHR3. These ions are usually disturbance in the spectra.
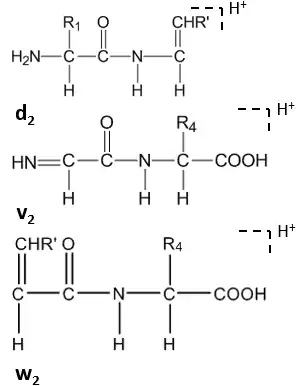
Further cleavage happens under high-energy CID at the side chain of C-terminal residues, forming dn, vn, wn-ions.[8]
Fragmentation rules summary
Most fragment ions are b- or y-ions. a-ions are also frequently seen by the loss of CO from b-ions.[9]
Satellite ions(wn, vn, dn-ions) are formed by high-energy CID.
Ser-, Thr-, Asp- and Glu-containing ions generate neutral molecular loss of water (-18).
Asn-, Gln-, Lys-, Arg-containing ions generate neutral molecular loss of ammonia (-17).
Neutral loss of ammonia from Arg leads to fragment ions (y-17) or (b-17) ions with higher abundance than their corresponding ions.
When C-terminus has a basic residue, the peptide generates (bn-1+18) ion.
A complementary b-y ion pair can be observed in multiply charged ions spectra. For this b-y ion pair, the sum of their subscripts is equal to the total number of amino acid residues in the unknown peptide.
If the C-terminus is Arg or Lys, y1-ion can be found in the spectrum to prove it.
Methods for peptide fragmentation
In low energy collision induced dissociation (CID), b- and y-ions are the main product ions. In addition, loss of ammonia (-17 Da) is observed in fragment with RKNQ amino acids in it. Loss of water (-18 Da) can be observed in fragment with STED amino acids in it. No satellite ions are shown in the spectra.
In high energy CID, all different types of fragment ions can be observed but no losses of ammonia or water.
In electron transfer dissociation (ETD) and electron capture dissociation (ECD), the predominant ions are c, y, z+1, z+2 and sometimes w ions.
For post source decay (PSD) in MALDI, a, b, y-ions are most common product ions.
Factors affecting fragmentation are the charge state (the higher charge state, the less energy is needed for fragmentation), mass of the peptide (the larger mass, the more energy is required), induced energy (higher energy leads to more fragmentation), primary amino acid sequence, mode of dissociation and collision gas.
Guidelines for interpretation
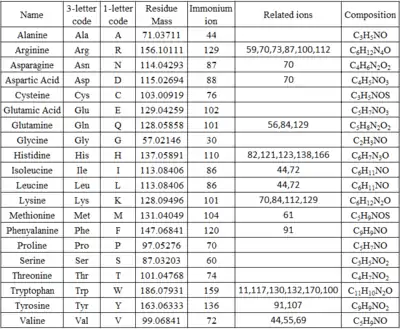
For interpretation,[14] first, look for single amino acid immonium ions (H2N+=CHR2). Corresponding immonium ions for amino acids are listed in Table 1. Ignore a few peaks at the high-mass end of the spectrum. They are ions that undergo neutral molecules losses (H2O, NH3, CO2, HCOOH) from [M+H]+ ions. Find mass differences at 28 Da since b-ions can form a-ions by loss of CO. Look for b2-ions at low-mass end of the spectrum, which helps to identify yn-2-ions too. Mass of b2-ions are listed in Table 2, as well as single amino acids that have equal mass to b2-ions.[15] The mass of b2-ion = mass of two amino acid residues + 1.
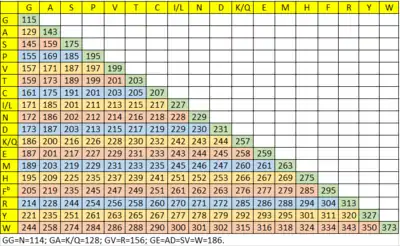
Identify a sequence ion series by the same mass difference, which matches one of the amino acid residue masses (see Table 1). For example, mass differences between an and an-1, bn and bn-1, cn and cn-1 are the same. Identify yn-1-ion at the high-mass end of the spectrum. Then continue to identify yn-2, yn-3... ions by matching mass differences with the amino acid residue masses (see Table 1). Look for the corresponding b-ions of the identified y-ions. The mass of b+y ions is the mass of the peptide +2 Da. After identifying the y-ion series and b-ion series, assign the amino acid sequence and check the mass. The other method is to identify b-ions first and then find the corresponding y-ions.
Algorithms and software
Manual de novo sequencing is labor-intensive and time-consuming. Usually algorithms or programs come with the mass spectrometer instrument are applied for the interpretation of spectra.
Development of de novo sequencing algorithms
An old method is to list all possible peptides for the precursor ion in mass spectrum, and match the mass spectrum for each candidate to the experimental spectrum. The possible peptide that has the most similar spectrum will have the highest chance to be the right sequence. However, the number of possible peptides may be large. For example, a precursor peptide with a molecular weight of 774 has 21,909,046 possible peptides. Even though it is done in the computer, it takes a long time.[17][18]
Another method is called "subsequencing", which instead of listing whole sequence of possible peptides, matches short sequences of peptides that represent only a part of the complete peptide. When sequences that highly match the fragment ions in the experimental spectrum are found, they are extended by residues one by one to find the best matching.[19][20][21][22]
In the third method, graphical display of the data is applied, in which fragment ions that have the same mass differences of one amino acid residue are connected by lines. In this way, it is easier to get a clear image of ion series of the same type. This method could be helpful for manual de novo peptide sequencing, but doesn't work for high-throughput condition.[23]
The fourth method, which is considered to be successful, is the graph theory. Applying graph theory in de novo peptide sequencing was first mentioned by Bartels.[24] Peaks in the spectrum are transformed into vertices in a graph called "spectrum graph". If two vertices have the same mass difference of one or several amino acids, a directed edge will be applied. The SeqMS algorithm,[25] Lutefisk algorithm,[26] Sherenga algorithm[27] are some examples of this type.
Deep Learning
More recently, deep learning techniques have been applied to solve the de novo peptide sequencing problem. The first breakthrough was DeepNovo, which adopted the convolutional neural network structure, achieved major improvements in sequence accuracy, and enabled complete protein sequence assembly without assisting databases[28] Subsequently, additional network structures, such as PointNet (PointNovo[29]), have been adopted to extract features from a raw spectrum. The de novo peptide sequencing problem is then framed as a sequence prediction problem. Given previously predicted partial peptide sequence, neural-network-based de novo peptide sequencing models will repeatedly generate the most probable next amino acid until the predicted peptide's mass matches the precursor mass. At inference time, search strategies such as beam search can be adopted to explore a larger search space while keeping the computational cost low. Comparing with previous methods, neural-network-based models have demonstrated significantly better accuracy and sensitivity.[28][29][30] Moreover, with a careful model design, deep-learning-based de novo peptide sequencing algorithms can also be fast enough to achieve real-time peptide de novo sequencing.[29] PEAKS software incorporates this neural network learning in their de novo sequencing algorithms.
Software packages
As described by Andreotti et al. in 2012,[31] Antilope is a combination of Lagrangian relaxation and an adaptation of Yen's k shortest paths. It is based on 'spectrum graph' method and contains different scoring functions, and can be comparable on the running time and accuracy to "the popular state-of-the-art programs" PepNovo and NovoHMM.
Grossmann et al.[32] presented AUDENS in 2005 as an automated de novo peptide sequencing tool containing a preprocessing module that can recognize signal peaks and noise peaks.
Lutefisk can solve de novo sequencing from CID mass spectra. In this algorithm, significant ions are first found, then determine the N- and C-terminal evidence list. Based on the sequence list, it generates complete sequences in spectra and scores them with the experimental spectrum. However, the result may include several sequence candidates that have only little difference, so it is hard to find the right peptide sequence. A second program, CIDentify, which is a modified version by Alex Taylor of Bill Pearson's FASTA algorithm, can be applied to distinguish those uncertain similar candidates.
Mo et al. presented the MSNovo algorithm in 2007 and proved that it performed "better than existing de novo tools on multiple data sets".[33] This algorithm can do de novo sequencing interpretation of LCQ, LTQ mass spectrometers and of singly, doubly, triply charged ions. Different from other algorithms, it applied a novel scoring function and use a mass array instead of a spectrum graph.
Fisher et al.[34] proposed the NovoHMM method of de novo sequencing. A hidden Markov model (HMM) is applied as a new way to solve de novo sequencing in a Bayesian framework. Instead of scoring for single symbols of the sequence, this method considers posterior probabilities for amino acids. In the paper, this method is proved to have better performance than other popular de novo peptide sequencing methods like PepNovo by a lot of example spectra.
PEAKS is a complete software package for the interpretation of peptide mass spectra. It contains de novo sequencing, database search, PTM identification, homology search and quantification in data analysis. Ma et al. described a new model and algorithm for de novo sequencing in PEAKS, and compared the performance with Lutefisk of several tryptic peptides of standard proteins, by the quadrupole time-of-flight (Q-TOF) mass spectrometer.[35]
PepNovo is a high throughput de novo peptide sequencing tool and uses a probabilistic network as scoring method. It usually takes less than 0.2 seconds for interpretation of one spectrum. Described by Frank et al., PepNovo works better than several popular algorithms like Sherenga, PEAKS, Lutefisk.[36] Now a new version PepNovo+ is available.
Chi et al. presented pNovo+ in 2013 as a new de novo peptide sequencing tool by using complementary HCD and ETD tandem mass spectra.[37] In this method, a component algorithm, pDAG, largely speeds up the acquisition time of peptide sequencing to 0.018s on average, which is three times as fast as the other popular de novo sequencing software.
As described by Jeong et al., compared with other do novo peptide sequencing tools, which works well on only certain types of spectra, UniNovo is a more universal tool that has a good performance on various types of spectra or spectral pairs like CID, ETD, HCD, CID/ETD, etc. It has a better accuracy than PepNovo+ or PEAKS. Moreover, it generates the error rate of the reported peptide sequences.[38]
Ma published Novor in 2015 as a real-time de novo peptide sequencing engine. The tool is sought to improve the de novo speed by an order of magnitude and retain similar accuracy as other de novo tools in the market. On a Macbook Pro laptop, Novor has achieved more than 300 MS/MS spectra per second.[39]
Pevtsov et al. compared the performance of the above five de novo sequencing algorithms: AUDENS, Lutefisk, NovoHMM, PepNovo, and PEAKS . QSTAR and LCQ mass spectrometer data were employed in the analysis, and evaluated by relative sequence distance (RSD) value, which was the similarity between de novo peptide sequencing and true peptide sequence calculated by a dynamic programming method. Results showed that all algorithms had better performance in QSTAR data than on LCQ data, while PEAKS as the best had a success rate of 49.7% in QSTAR data, and NovoHMM as the best had a success rate of 18.3% in LCQ data. The performance order in QSTAR data was PEAKS > Lutefisk, PepNovo > AUDENS, NovoHMM, and in LCQ data was NovoHMM > PepNovo, PEAKS > Lutefisk > AUDENS. Compared in a range of spectrum quality, PEAKS and NovoHMM also showed the best performance in both data among all 5 algorithms. PEAKS and NovoHMM had the best sensitivity in both QSTAR and LCQ data as well. However, no evaluated algorithms exceeded a 50% of exact identification for both data sets.[40]
Recent progress in mass spectrometers made it possible to generate mass spectra of ultra-high resolution . The improved accuracy, together with the increased amount of mass spectrometry data that are being generated, draws the interests of applying deep learning techniques to de novo peptide sequencing. In 2017 Tran et al. proposed DeepNovo, the first deep learning based de novo sequencing software. The benchmark analysis in the original publication demonstrated that DeepNovo outperformed previous methods, including PEAKS, Novor and PepNovo, by a significant margin. DeepNovo is implemented in python with the Tensorflow framework.[41] To represent a mass spectrum as a fixed-dimensional input to the neural-network, DeepNovo discretized each spectrum into a length 150,000 vector. This unnecessarily large spectrum representation, and the single-thread CPU usage in the original implementation, prevents DeepNovo from performing peptide sequencing in real time. To further improve efficiency of de novo peptide sequencing models, Qiao et al. proposed PointNovo in 2020. PointNovo is a python software implemented with the PyTorch framework [42] and it gets rid of the space consuming spectrum-vector-representation adopted by DeepNovo. Comparing with DeepNovo, PointNovo managed to achieve better accuracy and efficiency at the same time by directly representing a spectrum as a set of m/z and intensity pairs.
References
- ↑ Edman, P.; Begg, G. (March 1967). "A Protein Sequenator". European Journal of Biochemistry. 1 (1): 80–91. doi:10.1111/j.1432-1033.1967.tb00047.x. PMID 6059350.
- ↑ Webb-Robertson, B.-J. M.; Cannon, W. R. (20 June 2007). "Current trends in computational inference from mass spectrometry-based proteomics". Briefings in Bioinformatics. 8 (5): 304–317. doi:10.1093/bib/bbm023. PMID 17584764.
- ↑ Lu, Bingwen; Chen, Ting (March 2004). "Algorithms for de novo peptide sequencing using tandem mass spectrometry". Drug Discovery Today: BIOSILICO. 2 (2): 85–90. doi:10.1016/S1741-8364(04)02387-X.
- 1 2 Papayannopoulos, Ioannis A. (January 1995). "The interpretation of collision-induced dissociation tandem mass spectra of peptides". Mass Spectrometry Reviews. 14 (1): 49–73. Bibcode:1995MSRv...14...49P. doi:10.1002/mas.1280140104.
- ↑ Dass, Chhabil; Desiderio, Dominic M. (May 1987). "Fast atom bombardment mass spectrometry analysis of opioid peptides". Analytical Biochemistry. 163 (1): 52–66. doi:10.1016/0003-2697(87)90092-3. PMID 2887130.
- ↑ Yalcin, Talat; Csizmadia, Imre G.; Peterson, Michael R.; Harrison, Alex G. (March 1996). "The structure and fragmentation of B n (n≥3) ions in peptide spectra". Journal of the American Society for Mass Spectrometry. 7 (3): 233–242. doi:10.1016/1044-0305(95)00677-X. PMID 24203294.
- ↑ Tang, Xue-Jun; Boyd, Robert K.; Bertrand, M. J. (November 1992). "An investigation of fragmentation mechanisms of doubly protonated tryptic peptides". Rapid Communications in Mass Spectrometry. 6 (11): 651–657. Bibcode:1992RCMS....6..651T. doi:10.1002/rcm.1290061105. PMID 1467549.
- 1 2 3 Johnson, Richard S.; Martin, Stephen A.; Biemann, Klaus (December 1988). "Collision-induced fragmentation of (M + H)+ ions of peptides. Side chain specific sequence ions". International Journal of Mass Spectrometry and Ion Processes. 86: 137–154. Bibcode:1988IJMSI..86..137J. doi:10.1016/0168-1176(88)80060-0.
- 1 2 Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.]. ed.). Hoboken, N.J.: Wiley-Interscience. pp. 317–322. doi:10.1002/0470118490. ISBN 9780470118498.
- ↑ Dass, Chhabil (2001). Principles and practice of biological mass spectrometry. New York, NY [u.a.]: Wiley. ISBN 978-0-471-33053-0.
- ↑ Roepstorff, P; Fohlman, J (November 1984). "Proposal for a common nomenclature for sequence ions in mass spectra of peptides". Biomedical Mass Spectrometry. 11 (11): 601. doi:10.1002/bms.1200111109. PMID 6525415.
- ↑ McCloskey, James A., ed. (1990). Mass spectrometry. San Diego: Academic Press. pp. 886–887. ISBN 978-0121820947.
- ↑ Falick, A. M.; Hines, W. M.; Medzihradszky, K. F.; Baldwin, M. A.; Gibson, B. W. (November 1993). "Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry". Journal of the American Society for Mass Spectrometry. 4 (11): 882–893. doi:10.1016/1044-0305(93)87006-X. PMID 24227532.
- ↑ Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.]. ed.). Hoboken, N.J.: Wiley-Interscience. pp. 327–330. ISBN 9780470118498.
- ↑ Harrison, Alex G.; Csizmadia, Imre G.; Tang, Ting-Hua (May 2000). "Structure and fragmentation of b2 ions in peptide mass spectra". Journal of the American Society for Mass Spectrometry. 11 (5): 427–436. doi:10.1016/S1044-0305(00)00104-5. PMID 10790847. S2CID 24794690.
- ↑ Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.]. ed.). Hoboken, N.J.: Wiley-Interscience. p. 329. ISBN 9780470118498.
- ↑ Sakurai, T.; Matsuo, T.; Matsuda, H.; Katakuse, I. (August 1984). "PAAS 3: A computer program to determine probable sequence of peptides from mass spectrometric data". Biological Mass Spectrometry. 11 (8): 396–399. doi:10.1002/bms.1200110806.
- ↑ Hamm, C. W.; Wilson, W. E.; Harvan, D. J. (1986). "Peptide sequencing program". Bioinformatics. 2 (2): 115–118. doi:10.1093/bioinformatics/2.2.115. PMID 3450361.
- ↑ Biemann, K; Cone, C; Webster, BR; Arsenault, GP (5 December 1966). "Determination of the amino acid sequence in oligopeptides by computer interpretation of their high-resolution mass spectra". Journal of the American Chemical Society. 88 (23): 5598–606. doi:10.1021/ja00975a045. PMID 5980176.
- ↑ Ishikawa, K.; Niwa, Y. (July 1986). "Computer-aided peptide sequencing by fast atom bombardment mass spectrometry". Biological Mass Spectrometry. 13 (7): 373–380. doi:10.1002/bms.1200130709.
- ↑ Siegel, MM; Bauman, N (15 March 1988). "An efficient algorithm for sequencing peptides using fast atom bombardment mass spectral data". Biomedical & Environmental Mass Spectrometry. 15 (6): 333–43. doi:10.1002/bms.1200150606. PMID 2967723.
- ↑ Johnson, RS; Biemann, K (November 1989). "Computer program (SEQPEP) to aid in the interpretation of high-energy collision tandem mass spectra of peptides". Biomedical & Environmental Mass Spectrometry. 18 (11): 945–57. doi:10.1002/bms.1200181102. PMID 2620156.
- ↑ Scoble, Hubert A.; Biller, James E.; Biemann, Klaus (1987). "A graphics display-oriented strategy for the amino acid sequencing of peptides by tandem mass spectrometry". Fresenius' Zeitschrift für Analytische Chemie. 327 (2): 239–245. doi:10.1007/BF00469824. S2CID 97665981.
- ↑ Bartels, Christian (June 1990). "Fast algorithm for peptide sequencing by mass spectroscopy". Biological Mass Spectrometry. 19 (6): 363–368. doi:10.1002/bms.1200190607. PMID 24730078.
- ↑ Fernández-de-Cossío, J; Gonzalez, J; Besada, V (August 1995). "A computer program to aid the sequencing of peptides in collision-activated decomposition experiments". Computer Applications in the Biosciences. 11 (4): 427–34. doi:10.1093/bioinformatics/11.4.427. PMID 8521052.
- ↑ Taylor, JA; Johnson, RS (1997). "Sequence database searches via de novo peptide sequencing by tandem mass spectrometry". Rapid Communications in Mass Spectrometry. 11 (9): 1067–75. Bibcode:1997RCMS...11.1067T. doi:10.1002/(sici)1097-0231(19970615)11:9<1067::aid-rcm953>3.0.co;2-l. PMID 9204580.
- ↑ Dančík, Vlado; Addona, Theresa A.; Clauser, Karl R.; Vath, James E.; Pevzner, Pavel A. (October 1999). "Peptide Sequencing via Tandem Mass Spectrometry". Journal of Computational Biology. 6 (3–4): 327–342. CiteSeerX 10.1.1.128.2645. doi:10.1089/106652799318300. PMID 10582570.
- 1 2 Tran, Ngoc Hieu, etal. "De novo peptide sequencing by deep learning." Proceedings of the National Academy of Sciences 114.31 (2017): 8247-8252.
- 1 2 3 Qiao, Rui, et al. "Computationally instrument-resolution-independent de novo peptide sequencing for high-resolution devices." Nature Machine Intelligence 3.5 (2021): 420-425.
- ↑ Karunratanakul, Korrawe, et al. "Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework." Molecular & Cellular Proteomics 18.12 (2019): 2478-2491.
- ↑ Andreotti, S; Klau, GW; Reinert, K (2012). "Antilope--a Lagrangian relaxation approach to the de novo peptide sequencing problem". IEEE/ACM Transactions on Computational Biology and Bioinformatics. 9 (2): 385–94. arXiv:1102.4016. doi:10.1109/tcbb.2011.59. PMID 21464512. S2CID 593303.
- ↑ Grossmann, J; Roos, FF; Cieliebak, M; Lipták, Z; Mathis, LK; Müller, M; Gruissem, W; Baginsky, S (2005). "AUDENS: a tool for automated peptide de novo sequencing". Journal of Proteome Research. 4 (5): 1768–74. CiteSeerX 10.1.1.654.169. doi:10.1021/pr050070a. PMID 16212431.
- ↑ Mo, L; Dutta, D; Wan, Y; Chen, T (1 July 2007). "MSNovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry". Analytical Chemistry. 79 (13): 4870–8. doi:10.1021/ac070039n. PMID 17550227.
- ↑ Fischer, B; Roth, V; Roos, F; Grossmann, J; Baginsky, S; Widmayer, P; Gruissem, W; Buhmann, JM (15 November 2005). "NovoHMM: a hidden Markov model for de novo peptide sequencing". Analytical Chemistry. 77 (22): 7265–73. CiteSeerX 10.1.1.507.1610. doi:10.1021/ac0508853. PMID 16285674.
- ↑ Ma, Bin; Zhang, Kaizhong; Hendrie, Christopher; Liang, Chengzhi; Li, Ming; Doherty-Kirby, Amanda; Lajoie, Gilles (30 October 2003). "PEAKS: powerful software for peptidede novo sequencing by tandem mass spectrometry". Rapid Communications in Mass Spectrometry. 17 (20): 2337–2342. Bibcode:2003RCMS...17.2337M. doi:10.1002/rcm.1196. PMID 14558135.
- ↑ Frank, A; Pevzner, P (15 February 2005). "PepNovo: de novo peptide sequencing via probabilistic network modeling". Analytical Chemistry. 77 (4): 964–73. doi:10.1021/ac048788h. PMID 15858974.
- ↑ Chi, H; Chen, H; He, K; Wu, L; Yang, B; Sun, RX; Liu, J; Zeng, WF; Song, CQ; He, SM; Dong, MQ (1 February 2013). "pNovo+: de novo peptide sequencing using complementary HCD and ETD tandem mass spectra". Journal of Proteome Research. 12 (2): 615–25. doi:10.1021/pr3006843. PMID 23272783.
- ↑ Jeong, K; Kim, S; Pevzner, PA (15 August 2013). "UniNovo: a universal tool for de novo peptide sequencing". Bioinformatics. 29 (16): 1953–62. doi:10.1093/bioinformatics/btt338. PMC 3722526. PMID 23766417.
- ↑ Ma, Bin (30 June 2015). "Novor: Real-Time Peptide de Novo Sequencing Software". Journal of the American Society for Mass Spectrometry. 26 (11): 1885–1894. Bibcode:2015JASMS..26.1885M. doi:10.1007/s13361-015-1204-0. PMC 4604512. PMID 26122521.
- ↑ Pevtsov, S.; Fedulova, I.; Mirzaei, H.; Buck, C.; Zhang, X. (2006). "Performance Evaluation of Existing De Novo Sequencing Algorithms". Journal of Proteome Research. 5 (11): 3018–3028. doi:10.1021/pr060222h. PMID 17081053.
- ↑ Abadi, Martín, et al. "Tensorflow: A system for large-scale machine learning." 12th {USENIX} symposium on operating systems design and implementation ({OSDI} 16). 2016.
- ↑ Adam, et al. "Pytorch: An imperative style, high-performance deep learning library." Advances in neural information processing systems 32 (2019): 8026-8037.