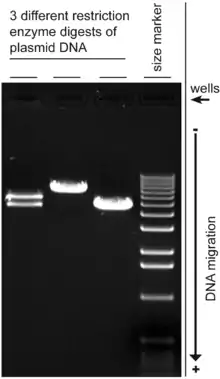
Agarose gel electrophoresis is a method of gel electrophoresis used in biochemistry, molecular biology, genetics, and clinical chemistry to separate a mixed population of macromolecules such as DNA or proteins in a matrix of agarose, one of the two main components of agar. The proteins may be separated by charge and/or size (isoelectric focusing agarose electrophoresis is essentially size independent), and the DNA and RNA fragments by length.[1] Biomolecules are separated by applying an electric field to move the charged molecules through an agarose matrix, and the biomolecules are separated by size in the agarose gel matrix.[2]
Agarose gel is easy to cast, has relatively fewer charged groups, and is particularly suitable for separating DNA of size range most often encountered in laboratories, which accounts for the popularity of its use. The separated DNA may be viewed with stain, most commonly under UV light, and the DNA fragments can be extracted from the gel with relative ease. Most agarose gels used are between 0.7–2% dissolved in a suitable electrophoresis buffer.
Properties of agarose gel
.jpg.webp)
Agarose gel is a three-dimensional matrix formed of helical agarose molecules in supercoiled bundles that are aggregated into three-dimensional structures with channels and pores through which biomolecules can pass.[3] The 3-D structure is held together with hydrogen bonds and can therefore be disrupted by heating back to a liquid state. The melting temperature is different from the gelling temperature, depending on the sources, agarose gel has a gelling temperature of 35–42 °C and a melting temperature of 85–95 °C. Low-melting and low-gelling agaroses made through chemical modifications are also available.
Agarose gel has large pore size and good gel strength, making it suitable as an anticonvection medium for the electrophoresis of DNA and large protein molecules. The pore size of a 1% gel has been estimated from 100 nm to 200–500 nm,[4][5] and its gel strength allows gels as dilute as 0.15% to form a slab for gel electrophoresis.[6] Low-concentration gels (0.1–0.2%) however are fragile and therefore hard to handle. Agarose gel has lower resolving power than polyacrylamide gel for DNA but has a greater range of separation, and is therefore used for DNA fragments of usually 50–20,000 bp in size. The limit of resolution for standard agarose gel electrophoresis is around 750 kb, but resolution of over 6 Mb is possible with pulsed field gel electrophoresis (PFGE).[7] It can also be used to separate large proteins, and it is the preferred matrix for the gel electrophoresis of particles with effective radii larger than 5–10 nm. A 0.9% agarose gel has pores large enough for the entry of bacteriophage T4.[6]
The agarose polymer contains charged groups, in particular pyruvate and sulphate.[8] These negatively charged groups create a flow of water in the opposite direction to the movement of DNA in a process called electroendosmosis (EEO), and can therefore retard the movement of DNA and cause blurring of bands. Higher concentration gels would have higher electroendosmotic flow. Low EEO agarose is therefore generally preferred for use in agarose gel electrophoresis of nucleic acids, but high EEO agarose may be used for other purposes. The lower sulphate content of low EEO agarose, particularly low-melting point (LMP) agarose, is also beneficial in cases where the DNA extracted from gel is to be used for further manipulation as the presence of contaminating sulphates may affect some subsequent procedures, such as ligation and PCR. Zero EEO agaroses however are undesirable for some applications as they may be made by adding positively charged groups and such groups can affect subsequent enzyme reactions.[9] Electroendosmosis is a reason agarose is used in preference to agar as the agaropectin component in agar contains a significant amount of negatively charged sulphate and carboxyl groups. The removal of agaropectin in agarose substantially reduces the EEO, as well as reducing the non-specific adsorption of biomolecules to the gel matrix. However, for some applications such as the electrophoresis of serum proteins, a high EEO may be desirable, and agaropectin may be added in the gel used.[10]
Migration of nucleic acids in agarose gel
Factors affecting migration of nucleic acid in gel
A number of factors can affect the migration of nucleic acids: the dimension of the gel pores (gel concentration), size of DNA being electrophoresed, the voltage used, the ionic strength of the buffer, and the concentration of intercalating dye such as ethidium bromide if used during electrophoresis.[11]
Smaller molecules travel faster than larger molecules in gel, and double-stranded DNA moves at a rate that is inversely proportional to the logarithm of the number of base pairs. This relationship however breaks down with very large DNA fragments, and separation of very large DNA fragments requires the use of pulsed field gel electrophoresis (PFGE), which applies alternating current from different directions and the large DNA fragments are separated as they reorient themselves with the changing field.[12]
For standard agarose gel electrophoresis, larger molecules are resolved better using a low concentration gel while smaller molecules separate better at high concentration gel. Higher concentration gels, however, require longer run times (sometimes days).
The movement of the DNA may be affected by the conformation of the DNA molecule, for example, supercoiled DNA usually moves faster than relaxed DNA because it is tightly coiled and hence more compact. In a normal plasmid DNA preparation, multiple forms of DNA may be present.[13] Gel electrophoresis of the plasmids would normally show the negatively supercoiled form as the main band, while nicked DNA (open circular form) and the relaxed closed circular form appears as minor bands. The rate at which the various forms move however can change using different electrophoresis conditions,[14] and the mobility of larger circular DNA may be more strongly affected than linear DNA by the pore size of the gel.[15]
Ethidium bromide which intercalates into circular DNA can change the charge, length, as well as the superhelicity of the DNA molecule, therefore its presence in gel during electrophoresis can affect its movement. For example, the positive charge of ethidium bromide can reduce the DNA movement by 15%.[12] Agarose gel electrophoresis can be used to resolve circular DNA with different supercoiling topology.[16]
DNA damage due to increased cross-linking will also reduce electrophoretic DNA migration in a dose-dependent way.[17][18]
The rate of migration of the DNA is proportional to the voltage applied, i.e. the higher the voltage, the faster the DNA moves. The resolution of large DNA fragments however is lower at high voltage. The mobility of DNA may also change in an unsteady field – in a field that is periodically reversed, the mobility of DNA of a particular size may drop significantly at a particular cycling frequency.[4] This phenomenon can result in band inversion in field inversion gel electrophoresis (FIGE), whereby larger DNA fragments move faster than smaller ones.
Migration anomalies
- "Smiley" gels - this edge effect is caused when the voltage applied is too high for the gel concentration used.[19]
- Overloading of DNA - overloading of DNA slows down the migration of DNA fragments.
- Contamination - presence of impurities, such as salts or proteins can affect the movement of the DNA.
Mechanism of migration and separation
The negative charge of its phosphate backbone moves the DNA towards the positively charged anode during electrophoresis. However, the migration of DNA molecules in solution, in the absence of a gel matrix, is independent of molecular weight during electrophoresis.[4][20] The gel matrix is therefore responsible for the separation of DNA by size during electrophoresis, and a number of models exist to explain the mechanism of separation of biomolecules in gel matrix. A widely accepted one is the Ogston model which treats the polymer matrix as a sieve. A globular protein or a random coil DNA moves through the interconnected pores, and the movement of larger molecules is more likely to be impeded and slowed down by collisions with the gel matrix, and the molecules of different sizes can therefore be separated in this sieving process.[4]
The Ogston model however breaks down for large molecules whereby the pores are significantly smaller than size of the molecule. For DNA molecules of size greater than 1 kb, a reptation model (or its variants) is most commonly used. This model assumes that the DNA can crawl in a "snake-like" fashion (hence "reptation") through the pores as an elongated molecule. A biased reptation model applies at higher electric field strength, whereby the leading end of the molecule become strongly biased in the forward direction and pulls the rest of the molecule along.[21] Real-time fluorescence microscopy of stained molecules, however, showed more subtle dynamics during electrophoresis, with the DNA showing considerable elasticity as it alternately stretching in the direction of the applied field and then contracting into a ball, or becoming hooked into a U-shape when it gets caught on the polymer fibres.[22][23]
General procedure
The details of an agarose gel electrophoresis experiment may vary depending on methods, but most follow a general procedure.
Casting of gel

The gel is prepared by dissolving the agarose powder in an appropriate buffer, such as TAE or TBE, to be used in electrophoresis.[12] The agarose is dispersed in the buffer before heating it to near-boiling point, but avoid boiling. The melted agarose is allowed to cool sufficiently before pouring the solution into a cast as the cast may warp or crack if the agarose solution is too hot. A comb is placed in the cast to create wells for loading sample, and the gel should be completely set before use.
The concentration of gel affects the resolution of DNA separation. The agarose gel is composed of microscopic pores through which the molecules travel, and there is an inverse relationship between the pore size of the agarose gel and the concentration – pore size decreases as the density of agarose fibers increases. High gel concentration improves separation of smaller DNA molecules, while lowering gel concentration permits large DNA molecules to be separated. The process allows fragments ranging from 50 base pairs to several mega bases to be separated depending on the gel concentration used.[24] The concentration is measured in weight of agarose over volume of buffer used (g/ml). For a standard agarose gel electrophoresis, a 0.8% gel gives good separation or resolution of large 5–10kb DNA fragments, while 2% gel gives good resolution for small 0.2–1kb fragments. 1% gels is often used for a standard electrophoresis.[25] High percentage gels are often brittle and may not set evenly, while low percentage gels (0.1-0.2%) are fragile and not easy to handle. Low-melting-point (LMP) agarose gels are also more fragile than normal agarose gel. Low-melting point agarose may be used on its own or simultaneously with standard agarose for the separation and isolation of DNA.[26] PFGE and FIGE are often done with high percentage agarose gels.
Loading of samples
Once the gel has set, the comb is removed, leaving wells where DNA samples can be loaded. Loading buffer is mixed with the DNA sample before the mixture is loaded into the wells. The loading buffer contains a dense compound, which may be glycerol, sucrose, or Ficoll, that raises the density of the sample so that the DNA sample may sink to the bottom of the well.[12] If the DNA sample contains residual ethanol after its preparation, it may float out of the well. The loading buffer also includes colored dyes such as xylene cyanol and bromophenol blue used to monitor the progress of the electrophoresis. The DNA samples are loaded using a pipette.
Electrophoresis

Agarose gel electrophoresis is most commonly done horizontally in a subaquaeous mode whereby the slab gel is completely submerged in buffer during electrophoresis. It is also possible, but less common, to perform the electrophoresis vertically, as well as horizontally with the gel raised on agarose legs using an appropriate apparatus.[27] The buffer used in the gel is the same as the running buffer in the electrophoresis tank, which is why electrophoresis in the subaquaeous mode is possible with agarose gel.
For optimal resolution of DNA greater than 2 kb in size in standard gel electrophoresis, 5 to 8 V/cm is recommended (the distance in cm refers to the distance between electrodes, therefore this recommended voltage would be 5 to 8 multiplied by the distance between the electrodes in cm).[14] Voltage may also be limited by the fact that it heats the gel and may cause the gel to melt if it is run at high voltage for a prolonged period, especially if the gel used is LMP agarose gel. Too high a voltage may also reduce resolution, as well as causing band streaking for large DNA molecules. Too low a voltage may lead to broadening of band for small DNA fragments due to dispersion and diffusion.[28]
Since DNA is not visible in natural light, the progress of the electrophoresis is monitored using colored dyes. Xylene cyanol (light blue color) comigrates large DNA fragments, while Bromophenol blue (dark blue) comigrates with the smaller fragments. Less commonly used dyes include Cresol Red and Orange G which migrate ahead of bromophenol blue. A DNA marker is also run together for the estimation of the molecular weight of the DNA fragments. Note however that the size of a circular DNA like plasmids cannot be accurately gauged using standard markers unless it has been linearized by restriction digest, alternatively a supercoiled DNA marker may be used.
Staining and visualization

DNA as well as RNA are normally visualized by staining with ethidium bromide, which intercalates into the major grooves of the DNA and fluoresces under UV light. The intercalation depends on the concentration of DNA and thus, a band with high intensity will indicate a higher amount of DNA compared to a band of less intensity.[12] The ethidium bromide may be added to the agarose solution before it gels, or the DNA gel may be stained later after electrophoresis. Destaining of the gel is not necessary but may produce better images. Other methods of staining are available; examples are MIDORI Green, SYBR Green, GelRed, methylene blue, brilliant cresyl blue, Nile blue sulphate, and crystal violet.[29] SYBR Green, GelRed and other similar commercial products are sold as safer alternatives to ethidium bromide as it has been shown to be mutagenic in Ames test, although the carcinogenicity of ethidium bromide has not actually been established. SYBR Green requires the use of a blue-light transilluminator. DNA stained with crystal violet can be viewed under natural light without the use of a UV transilluminator which is an advantage, however it may not produce a strong band.
When stained with ethidium bromide, the gel is viewed with an ultraviolet (UV) transilluminator. The UV light excites the electrons within the aromatic ring of ethidium bromide, and once they return to the ground state, light is released, making the DNA and ethidium bromide complex fluoresce.[12] Standard transilluminators use wavelengths of 302/312-nm (UV-B), however exposure of DNA to UV radiation for as little as 45 seconds can produce damage to DNA and affect subsequent procedures, for example reducing the efficiency of transformation, in vitro transcription, and PCR.[30] Exposure of DNA to UV radiation therefore should be limited. Using a higher wavelength of 365 nm (UV-A range) causes less damage to the DNA but also produces much weaker fluorescence with ethidium bromide. Where multiple wavelengths can be selected in the transilluminator, shorter wavelength can be used to capture images, while longer wavelength should be used if it is necessary to work on the gel for any extended period of time.
The transilluminator apparatus may also contain image capture devices, such as a digital or polaroid camera, that allow an image of the gel to be taken or printed.
For gel electrophoresis of protein, the bands may be visualised with Coomassie or silver stains.

Downstream procedures
The separated DNA bands are often used for further procedures, and a DNA band may be cut out of the gel as a slice, dissolved and purified. Contaminants however may affect some downstream procedures such as PCR, and low melting point agarose may be preferred in some cases as it contains fewer of the sulphates that can affect some enzymatic reactions. The gels may also be used for blotting techniques.
Buffers
In general, the ideal buffer should have good conductivity, produce less heat and have a long life.[31] There are a number of buffers used for agarose electrophoresis; common ones for nucleic acids include Tris/Acetate/EDTA (TAE) and Tris/Borate/EDTA (TBE). The buffers used contain EDTA to inactivate many nucleases which require divalent cation for their function. The borate in TBE buffer can be problematic as borate can polymerize, and/or interact with cis diols such as those found in RNA. TAE has the lowest buffering capacity, but it provides the best resolution for larger DNA. This means a lower voltage and more time, but a better product.
Many other buffers have been proposed, e.g. lithium borate (LB), iso electric histidine, pK matched goods buffers, etc.; in most cases the purported rationale is lower current (less heat) and or matched ion mobilities, which leads to longer buffer life. Tris-phosphate buffer has high buffering capacity but cannot be used if DNA extracted is to be used in phosphate sensitive reaction. LB is relatively new and is ineffective in resolving fragments larger than 5 kbp; However, with its low conductivity, a much higher voltage could be used (up to 35 V/cm), which means a shorter analysis time for routine electrophoresis. As low as one base pair size difference could be resolved in 3% agarose gel with an extremely low conductivity medium (1 mM lithium borate).[32]
Other buffering system may be used in specific applications, for example, barbituric acid-sodium barbiturate or Tris-barbiturate buffers may be used for in agarose gel electrophoresis of proteins, for example in the detection of abnormal distribution of proteins.[33]
Applications
- Estimation of the size of DNA molecules following digestion with restriction enzymes, e.g., in restriction mapping of cloned DNA.
- Estimation of the DNA concentration by comparing the intensity of the nucleic acid band with the corresponding band of the size marker.[34]
- Analysis of products of a polymerase chain reaction (PCR), e.g., in molecular genetic diagnosis or genetic fingerprinting
- Separation of DNA fragments for extraction and purification.
- Separation of restricted genomic DNA prior to Southern transfer, or of RNA prior to Northern transfer.
- Separation of proteins, for example, screening of protein abnormalities in clinical chemistry.[35]
Agarose gels are easily cast and handled compared to other matrices and nucleic acids are not chemically altered during electrophoresis. Samples are also easily recovered. After the experiment is finished, the resulting gel can be stored in a plastic bag in a refrigerator.
Electrophoresis is performed in buffer solutions to reduce pH changes due to the electric field, which is important because the charge of DNA and RNA depends on pH, but running for too long can exhaust the buffering capacity of the solution. Further, different preparations of genetic material may not migrate consistently with each other, for morphological or other reasons.
See also
References
- ↑ Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV (December 2003). "Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104". The Journal of Biological Chemistry. 278 (49): 49636–43. doi:10.1074/jbc.M307996200. PMID 14507919.
- ↑ Sambrook J, Russel DW (2001). Molecular Cloning: A Laboratory Manual 3rd Ed. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY.
- ↑ Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. Vol. 1 (3rd ed.). p. 5.4. ISBN 978-0-87969-577-4.
- 1 2 3 4 Zimm BH, Levene SD (May 1992). "Problems and prospects in the theory of gel electrophoresis of DNA" (PDF). Quarterly Reviews of Biophysics. 25 (2): 171–204. doi:10.1017/s0033583500004662. PMID 1518924. S2CID 27976751.
- ↑ Jean-Louis Viovy (2000). "Electrophoresis of DNA and other polyelectrolytes: Physical mechanisms". Reviews of Modern Physics. 72 (3): 813–872. Bibcode:2000RvMP...72..813V. doi:10.1103/RevModPhys.72.813.
- 1 2 Philip Serwer (1983). "Agarose gels: Properties and use for electrophoresis". Electrophoresis. 4 (6): 375–382. doi:10.1002/elps.1150040602. S2CID 97819634.
- ↑ Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. Vol. 1 (3rd ed.). p. 5.2–5.3. ISBN 978-0-87969-577-4.
- ↑ "Appendix B: Agarose Physical Chemistry" (PDF). Lonza Group. Archived (PDF) from the original on 2022-10-09.
- ↑ Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. Vol. 1 (3rd ed.). p. 5.7. ISBN 978-0-87969-577-4.
- ↑ Keren, David (26 September 2003). Protein Electrophoresis in Clinical Diagnosis. CRC Press. pp. 7–8. ISBN 978-0340812136.
- ↑ G. Lucotte; F. Baneyx (1993). Introduction to Molecular Cloning Techniques. Wiley-Blackwell. p. 32. ISBN 978-0471188490.
- 1 2 3 4 5 6 Lee PY, Costumbrado J, Hsu CY, Kim YH (April 2012). "Agarose gel electrophoresis for the separation of DNA fragments". Journal of Visualized Experiments (62). doi:10.3791/3923. PMC 4846332. PMID 22546956.
- ↑ Richard R. Sinden (1994-11-24). DNA Structure and Function. Academic Press Inc. p. 97. ISBN 978-0126457506.
- 1 2 Joseph Sambrook; David Russell. "Chapter 5, protocol 1". Molecular Cloning - A Laboratory Manual. Vol. 1 (3rd ed.). p. 5.5–5.6. ISBN 978-0-87969-577-4.
- ↑ Aaij C, Borst P (May 1972). "The gel electrophoresis of DNA". Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis. 269 (2): 192–200. doi:10.1016/0005-2787(72)90426-1. PMID 5063906.
- ↑ Donald Voet; Judith G. Voet (1995). Biochemistry (2nd ed.). John Wiley & Sons. pp. 877–878. ISBN 978-0471586517.
- ↑ Blasiak J, Trzeciak A, Malecka-Panas E, Drzewoski J, Wojewódzka M (August 2000). "In vitro genotoxicity of ethanol and acetaldehyde in human lymphocytes and the gastrointestinal tract mucosa cells". Toxicology in Vitro. 14 (4): 287–95. doi:10.1016/S0887-2333(00)00022-9. PMID 10906435.
- ↑ Lu Y, Morimoto K (July 2009). "Is habitual alcohol drinking associated with reduced electrophoretic DNA migration in peripheral blood leukocytes from ALDH2-deficient male Japanese?". Mutagenesis. 24 (4): 303–8. doi:10.1093/mutage/gep008. PMID 19286920.
- ↑ G. Lucotte; F. Baneyx (1993). Introduction to Molecular Cloning Techniques. Wiley-Blackwell. p. 41. ISBN 978-0471188490.
- ↑ Robert W. Old; Sandy B. Primrose (1994-09-27). Principle of Gene Manipulation - An Introduction to Genetic Engineering (5th ed.). Blackwell Scientific. p. 9. ISBN 9780632037124.
- ↑ Li Zhu; Hong Wang (2009-03-02). "Chapter 4 - Genetic Analysis in Miniaturized Electrophoresis Systems". In Tian, Wei-Cheng; Finehout, Erin (eds.). Microfluidics for Biological Applications. Springer. p. 125. ISBN 978-0-387-09480-9.
- ↑ Smith SB, Aldridge PK, Callis JB (January 1989). "Observation of individual DNA molecules undergoing gel electrophoresis". Science. 243 (4888): 203–6. Bibcode:1989Sci...243..203S. doi:10.1126/science.2911733. PMID 2911733.
- ↑ Schwartz DC, Koval M (April 1989). "Conformational dynamics of individual DNA molecules during gel electrophoresis". Nature. 338 (6215): 520–2. Bibcode:1989Natur.338..520S. doi:10.1038/338520a0. PMID 2927511. S2CID 4249063.
- ↑ Magdeldin, Sameh (2012). Gel Electrophoresis. InTech. pp. 35–40.
- ↑ "Agarose gel electrophoresis (basic method)". Biological Protocols. Retrieved 23 August 2011.
- ↑ Fotadar U, Shapiro LE, Surks MI (February 1991). "Simultaneous use of standard and low-melting agarose for the separation and isolation of DNA by electrophoresis". BioTechniques. 10 (2): 171–2. PMID 2059440.
- ↑ David Freifelder (1982). Physical Biochemistry: Applications to Biochemistry and Molecular Biology (2nd ed.). WH Freeman. pp. 292–293. ISBN 978-0716714446.
- ↑ "Section III: Loading and Running DNA in Agarose Gels" (PDF). Lonza Group. Archived (PDF) from the original on 2022-10-09.
- ↑ "DNA revealed" (PDF). National Centre for Biotechnology education. University of Reading. Archived from the original (PDF) on 2012-03-04.
- ↑ Gründemann D, Schömig E (November 1996). "Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light". BioTechniques. 21 (5): 898–903. doi:10.2144/96215rr02. PMID 8922632.
- ↑ Sameh Magdeldin, ed. (2012). Gel electrophoresis – Principles and Basics. InTech. ISBN 978-953-51-0458-2.
- ↑ Brody JR, Kern SE (October 2004). "History and principles of conductive media for standard DNA electrophoresis" (PDF). Analytical Biochemistry. 333 (1): 1–13. doi:10.1016/j.ab.2004.05.054. PMID 15351274. Archived from the original (PDF) on 24 December 2012.
- ↑ Jeppsson JO, Laurell CB, Franzén B (April 1979). "Agarose gel electrophoresis". Clinical Chemistry. 25 (4): 629–38. doi:10.1093/clinchem/25.4.629. PMID 313856.
- ↑ Mészáros, Éva (2021). "Determine the concentration and purity of nucleic acids". INTEGRA Biosciences.
- ↑ Giot, Jean-Francois (2010). "Agarose gel electrophoresis: Application in Clinical Chemistry" (PDF). Journal of Medical Biochemistry. 29: 9–14. doi:10.2478/v10011-009-0033-8. S2CID 94646249. Archived (PDF) from the original on 2022-10-09.
External links
- How to run a DNA or RNA gel
- Animation of gel analysis of DNA restriction fragments
- Video and article of agarose gel electrophoresis
- Step by step photos of running a gel and extracting DNA
- Drinking straw electrophoresis!
- A typical method from wikiversity
- Building a gel electrophoresis chamber