Gene deserts are regions of the genome that are devoid of protein-coding genes. Gene deserts constitute an estimated 25% of the entire genome, leading to the recent interest in their true functions.[1] Originally believed to contain inessential and “junk” DNA due to their inability to create proteins, gene deserts have since been linked to several vital regulatory functions, including distal enhancing and conservatory inheritance. Thus, an increasing number of risks that lead to several major diseases, including a handful of cancers, have been attributed to irregularities found in gene deserts. One of the most notable examples is the 8q24 gene region, which, when affected by certain single nucleotide polymorphisms, lead to a myriad of diseases. The major identifying factors of gene deserts lay in their low GpC content and their relatively high levels of repeats, which are not observed in coding regions. Recent studies have even further categorized gene deserts into variable and stable forms; regions are categorized based on their behavior through recombination and their genetic contents. Although current knowledge of gene deserts is rather limited, ongoing research and improved techniques are beginning to open the doors for exploration on the various important effects of these noncoding regions.
History
Although the possibility of function in gene deserts was predicted as early as the 1960s, genetic identification tools were unable to uncover any specific characteristics of the long noncoding regions, other than that no coding occurred in those regions.[2] Before the completion of the human genome in 2001 through the Human Genome Project, most of the early associative gene comparisons relied on the belief that essential housekeeping genes were clustered in the same areas of the genome for ease of access and tight regulation. This belief later constructed a hypothesis that gene deserts are therefore previous regulatory sequences that are highly linked (and hence do not undergo recombination), but have had substitutions between them over time.[2][3] These substitutions could cause tightly conserved genes to separate over time, thus forming regions of nonsense codes with a few essential genes. However, uncertainty due to differential gene conservation rates in different portions of chromosomes prevented accurate identification. Later associations were remodeled when regulatory sequences were associated with transcription factors, leading to the birth of large-scale genome-wide mapping. Thus began the hunt for the contents and functions of gene deserts. Recent advancements in the screening of chromatin signatures on chromosomes (for instance, chromosome conformation capture, also known as 3C) have allowed the confirmation of the long-range gene activation model, which postulates that there are indeed physical links between regulatory enhancers and their target promoters.[2] Research on gene deserts, although centralized on human genetics, has also been applied to mice, various birds, and Drosophila melanogaster.[4][5] Although conservation is variable among selected species’ genomes, orthologous gene deserts function similarly. Thus, the prevailing the contention of gene deserts is that these noncoding sequences harbor active and important regulatory elements.
Possible functions
One study focused on a regulatory archipelago, a region with “islands” of coding sequences surrounded by vast noncoding regions. The study, which explored the effects of regulation on the hox genes, initially focused on two enhancer sequences, GCR and Prox, which are located 200 basepairs and 50 basepairs respectively upstream of the Hox D locus.[5] To manipulate the region, the study inverted the two enhancer sequences and discovered no major effects on the transcription of the Hox D gene, even though the two sequences were the closest sequences to the gene. Thus, the turned to the gene desert that flanked the GCR sequence upstream and found 5 regulatory islands within it that could regulate the gene. To select the most likely candidate, the study then applied several individual and multiple deletions to the five islands to observe the effects. These varied deletions only resulted in minor effects including physical abnormalities or a few missing digits.
When the experiment was taken a step further and applied a deletion of the entire 830 kilobase gene desert, the functionality of the entire Hox D locus was rendered inactive.[5] This indicates that the neighboring gene desert, as an entire 830 kilobase unit (including the five island sequences within it), serves as an important regulator of a single gene that spans merely 50 kilobases. Therefore, these results hinted at the regulatory effects of flanking gene deserts. This study was supported by a later observation through a comparison between fluorescence in situ hybridization and chromosome conformation capture which discovered that the Hox D locus was the most decondensed portion in the region. This meant that it had relatively higher activity in comparison to the flanking gene deserts.[6] Hence, the Hox D could be regulated by specific nearby enhancer sequences that were not expressed in unison. However, this does caution that proximity is inaccurate when either analytical method is used.[6] Thus, associations between regulatory gene deserts and their target promoters seem to have variable distances and are not required to act as borders.

The variability in distance demonstrates that distance may be another important factor that is determined by gene deserts. For instance, distal enhancers may interact with their target promoters through looping interactions which must act over a certain distance.[7] Thus, proximity is not an accurate predictor of enhancers: enhancers do not need to border their target sequence to regulate them. While this leads to a variation in distances, the average distance between transcription start sites and the interaction complex mediated by their enhancer elements is 120 kilobases upstream of the start site.[7] Gene deserts may play a role in constructing this distance to allow maximal looping to occur. Given that the mechanism of enhancer complex formation is a fairly simply regulated mechanism (the structures that are recruited into the enhancing complex have various regulatory controls that control construction), more than 50% of promoters have several long-range interactions. Certain core genes even have up to 20 possible enhancing interactions. There is a curious bias for complexes to form only upstream of the promoters.[7] Thus, given the correlation that many regulatory gene deserts appear upstream of their target promoters, it is possible that the more immediate role that gene deserts play is in long-range regulation of key sequences. As the ideal formation of enhancer interactions requires specific constructs, a possible side-product of the regulatory roles of gene deserts may be the conservation of genes: to retain the specific lengths of loops and order of regulating genes hidden in gene deserts, certain portions of gene deserts are more highly conserved than others when passing through inheritance events. These conserved noncoding sequences (CNS) are directly associated with syntenic inheritance in all vertebrates.[8] Thus, the presence of these CNSs could serve to conserve of large regions of genes. Although distance may vary in regulatory gene deserts, distance appears to have an upper limit in conservative gene deserts. CNSs were initially thought to occur close to their conserved genes: earlier estimates placed most CNSs in proximity of gene sequences.[8] However, the expansion of genetic data has revealed that several CNSs reside up to 2.5 megabases from their target genes, with the majority of CNSs falling between 1 and 2 megabases. This range, which was measured for the human genome, is varied among different species. For instance, in comparison to humans, the Fugu fish has a smaller range, with an estimated maximum distance of a few hundred kilobases. Regardless of the difference in lengths, CNSs work in similar methods in both species.[8] Thus, as functions differ between gene deserts, so do their contents.
Stable and variable gene deserts

Certain gene deserts are heavy regulators, while others may be deleted without any effect. As a possible classification, gene deserts can be broken down into two subtypes: stable and variable.[1] Stable gene deserts have fewer repeats and have relatively higher Guanine to Cytosine (GpC) content than observed in variable gene deserts.
Guanine and cytosine content is indicative of protein-coding functionality. For example, in a study on chromosomes 2 and 4, which have been linked to several genetic diseases, there were elevated GpC content in certain regions.[9] Mutations in these GC-rich regions caused a variety of diseases, revealing the necessary integrity of these genes. High density CpG regions serve as regulatory regions for DNA methylation.[10] Therefore, essential coding genes should be represented by high-CpG regions. In particular, regions with high GC content should tend to have high densities of genes that are devoted mainly to the essential housekeeping and tissue specific processes.[11] These processes would require the most protein production to express functionality. Stable gene deserts, which have higher levels of GC content, should therefore contain the essential enhancer sequences. This could determine the conservatory functions of stable gene deserts.
On the other hand, approximately 80% of gene deserts have low GpC contents, indicating that they have very few essential genes.[9] Thus, the majority of gene deserts are variable gene deserts, which may have alternate functions. One prevalent theory regarding the origins of gene deserts postulates that gene deserts are accumulations of essential genes that act as a distance.[1][10] This may hold true, as given the low numbers of essential genes within them, these regions would have been less conserved. As a result, due to the prevalence of cytosine to thymine conversions, the most common SNP, would cause a gradual separation between the few essential genes within variable gene deserts. These essential sequences would have been maintained and conserved, leading to small regions of high density that regulate at a distance.[10] GC content is therefore indication for the presence of coding or regulatory processes in DNA.
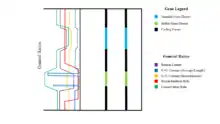
While stable gene deserts have higher GC content, this relative value is only an average. Within stable gene deserts, although the ends contain very high levels of GC content, the main bulk of the DNA contains even less GC content than observed in variable gene deserts. This indicates that there are very few highly conserved regions in stable gene deserts that do not recombine, or do so at very low rates.[9] Given that the ends of the stable gene deserts have particularly high levels of GC contents, these sequences must be extremely conserved. This conservation may in turn cause the flanking genes to also have higher conservation rates. Thus, stable genes should be directly linked to at least one of their flanking genes and cannot be separated from coding sequences by recombination events.[1] Most gene deserts appear to cluster in pairs around a small number of genes. This clustering creates long loci that have very low gene density; small regions with high numbers of genes are surrounded by long stretches of gene deserts, creating a low gene average. Therefore, the minimized probability of recombination events in these long loci creates syntenic blocks that are inherited together over time.[1] These syntenic blocks can be conserved for very long periods of time, preventing loss of essential material, even while the distance between essential genes may grow in time.
Although this effect should theoretically be amplified through the even lower GC-content in variable gene deserts (thereby truly minimalizing gene density), the gene conservation rates in variable gene deserts are even lower than observed in stable gene deserts—in fact, the rate is far lower than the rest of the genome. A possible explanation for this phenomenon is that variable gene deserts may be recently evolved regions that have not yet been fixed into stable gene deserts.[1] Therefore, shuffling may still occur before stabilizing regions within the variable gene deserts begin to cluster as whole units. There are a few exceptions to this minimal rate of conservation, as a few GC gene deserts are subjected to hypermethylation, which greatly reduces the accessibility to the DNA, thus effectively protecting the region from recombination.[11] However, these occur rarely in observation.
Although stable and variable gene deserts differ in content and function, both wield conservatory abilities. It is possible that since most variable gene deserts have regulatory elements that can act at a distance, conservation of the entire gene desert into a sytenic locus would not have been necessary, so long as these regulatory elements themselves were conserved as units. Given the particularly low levels of GC content, the regulatory elements would therefore be in a minimal gene density situation as observed similarly in flanking stable gene deserts, with the same effect. Thus, both types of gene deserts serve to retain essential genes within the genome.
Genetic diseases
The conservative nature of gene deserts confirms that these stretches of noncoding bases are essential to proper functioning. Indeed, a wide range of studies on irregularities in the noncoding genes discovered several associations to genetic diseases. One of the most studied gene deserts is the 8q24 region. Early genome wide association studies were focused on the 8q24 region (residing on chromosome 8) due to the abnormally high rates of SNPs that seem to occur in the region. These studies found that the region was linked to increased risks for a variety of cancers, notably in the prostate, breast, ovaries, colonic, and pancreas.[12][13] Using inserts of the gene desert into bacterial artificial chromosomes, one study was able to produce enhancer activity in certain regions, which were isolated via cloning systems.[14] This study successfully identified an enhancer sequence hidden in the region. Within this enhancer sequence, an SNP that conferred risk for prostate cancer, labeled SNP s6983267, was discovered in diseased mice. However, the 8q24 region is not solely limited to conferred risks of prostate cancer. A study in 2008 screened human subjects (and controls) with variations in the gene desert region, discovering five different regions that conferred different risks when affected by different SNPs.[12] This study used identified SNP markers in the gene desert to identify risk conference from each of the regions to a specific tissue expression. Although these risks were successfully linked to various forms of cancer, Ghoussaini, M., et al. note their uncertainty in whether the SNPs functioned merely as markers or were the direct causants of the cancers.
These varied effects occur due to the different interactions between the SNPs in this region and MYC promoters of different organs. The MYC promoter, which is located at a short distance downstream of the 8q24 region, is perhaps the most studied oncogene due to its association with a myriad of diseases.[13] Normal functioning of the MYC promoter ensures that cells divide regularly. The study postulates that the 8q region, which underwent a chromosomal translocation in humans, could have moved an essential enhancer for the MYC promoter.[13] This areas around this region could have been subjected to recombination that may have hidden the essential MYC enhancer within the gene desert through time, although its enhancing effects are still very much retained. This analysis stems from disease associations observed in several mice species where this region is retained at proximity to the MYC promoter.[13] Thus, the 8q24 gene desert should have been somewhat linked to the MYC promoter. The desert resembles a stable gene desert that has had very little recombination after the translocation event. Thus, a potential hypothesis is that SNPs affecting this region disrupt the important tissue-specific genes with the stable gene desert, which could explain the risks of cancer in various tissue forms. This effect of hidden enhancer elements can also be observed in other locations in the genome. For instance, SNPs in the 5p13.1 deregulate the PTGER4 coding region, leading to Crohn's Disease.[15] Another affected region in the 9p21 gene desert causes several coronary artery diseases.[16] However, none of these risk-conferring gene deserts seem to be affected as much as the 8q24 regions. Current studies are still unsure about the SNP-affected processes in the 8q24 region that result in particularly amplified responses to the MYC promoter. With the aid of a more accessible population and more specific markers for genome wide association mapping, an increasing number of risk alleles are now being marked in gene deserts, where small, isolated, and seemingly-unimportant regions of genes may moderate important genes.
A caveat
It is crucial to note that although most of the gene deserts explored here are essential, it could be that the majority of the contents in gene deserts are still likely to be inessential and disposable. Naturally, this is not to say that the roles that gene deserts play are inessential or unimportant, rather than their functions may include buffering effects. An example of essential gene deserts with inessential DNA content are the telomeres that protect the ends of genomes. Telomeres can be categorized as true gene deserts, given that they solely contain repeats of TTAGGG (in humans) and do not have apparent protein-coding functions. Without these telomeres, human genomes would be severely mutated within a fixed number of cell cycles. On the other hand, since telomeres do not code for proteins, their loss ensures that there is no effect in important processes. Therefore, the term “junk” DNA should no longer be applied to any region of the genome; every portion of the genome should play a role in protecting, regulating, or repairing the protein coding regions that determine the functions of life. Although there is still much to learn about the nooks and crannies of the immense (yet limited) human genome, with the aid of various new technologies and the synthesis of the full human genome, we may perhaps unravel a great collection of secrets in the approaching years about the marvels of our genetic code.
See also
References
- 1 2 3 4 5 6 Ovcharenko, Ivan; et al. (December 2004). "Evolution and Functional Classification of Vertebrate Gene Deserts". Genome Research. 15 (1): 137–145. doi:10.1101/gr.3015505. PMC 540279. PMID 15590943.
- 1 2 3 Montavon, Thomas; Duboule, Denis (July 2012). "Landscapes and Archipelagos: Spatial Organization of Gene Regulation in Vertebrates". Trends in Cell Biology. 22 (7): 347–354. doi:10.1016/j.tcb.2012.04.003. PMID 22560708.
- ↑ Taylor, James (19 April 2005). "Clues to Function in Gene Deserts". Trends in Biotechnology. 23 (6): 269–271. doi:10.1016/j.tibtech.2005.04.003. PMID 15922077.
- ↑ Cooper, Monica; Kennison, James (August 2011). "Molecular Genetic Analyses of Polytene Chromosome Region 72A-D in Drosophila melanogaster Reveal a Gene Desert in 72D". PLOS ONE. 6 (8): e23509. Bibcode:2011PLoSO...623509C. doi:10.1371/journal.pone.0023509. PMC 3154481. PMID 21853143.
- 1 2 3 Montavon, Thomas; et al. (23 November 2011). "A Regulatory Archipelago Controls Hox Genes Transcription in Digits". Cell. 147 (5): 1132–1145. doi:10.1016/j.cell.2011.10.023. PMID 22118467.
- 1 2 Williamson, Iain; et al. (30 October 2014). "Spatial Genome Organization: Contrasting Views From Chromosome Conformation and Fluorescence in situ Hybridization". Genes & Development. 28 (24): 2778–2791. doi:10.1101/gad.251694.114. PMC 4265680. PMID 25512564.
- 1 2 3 Sanyal, Amartya; Lajoie, Bryan; Jain, Gaurav; Dekker, Job (6 September 2012). "The Long-Range Interaction Landscape of Gene Promoters". Nature. 489 (7414): 109–113. Bibcode:2012Natur.489..109S. doi:10.1038/nature11279. PMC 3555147. PMID 22955621.
- 1 2 3 Woolfe, Adam; Elgar, Greg (2008). "Organization of Conserved Elements Near Key Developmental Regulators in Vertebrate Genomes". Advances in Genetics. 61: 307–38. doi:10.1016/S0065-2660(07)00012-0. PMID 18282512.
- 1 2 3 Hillier, LaDeana; et al. (7 April 2005). "Generation and Annotation of the DNA Sequences of Human Chromosomes 2 and 4". Nature. 434 (7034): 724–731. Bibcode:2005Natur.434..724H. doi:10.1038/nature03466. PMID 15815621.
- 1 2 3 Skinner, Michael; Bosagna, Carlos (2014). "Role of CpG Deserts in the Epigenetic Transgenerational Inheritance of Differential DNA Methylation Regions". BMC Genomics. 15 (692): 692. doi:10.1186/1471-2164-15-692. PMC 4149044. PMID 25142051.
- 1 2 Zaghloul, Lamia; et al. (11 July 2014). "Large Replication Skew Domains Delimit GC-poor Gene Deserts in Human". Computational Biology and Chemistry. 53: 153–165. doi:10.1016/j.compbiolchem.2014.08.020. PMID 25224847.
- 1 2 Ghoussaini, Maya; et al. (2 July 2008). "Multiple Loci with Different Cancer Specificities Within the 8q24 Gene Desert". Brief Communications. 100 (13).
- 1 2 3 4 Huppi, Konrad; Pitt, Jason; Wahlberg, Brady; Caplen, Natasha (April 2012). "The 8q24 Gene Desert: An Oasis of Noncoding Transcriptional Activity". Frontiers in Genetics. 3: 69. doi:10.3389/fgene.2012.00069. PMC 3339310. PMID 22558003.
- ↑ Wasserman, Nora; Aneas, Ivy; Nobrega, Marcelo (3 June 2010). "An 8q24 Gene Desert Variant Associated with Prostate Cancer Risk Confers Differential in vivo Activity to a MYC Enhancer". Genome Research. 20 (9): 1191–1197. doi:10.1101/gr.105361.110. PMC 2928497. PMID 20627891.
- ↑ Libioulle, Cecile; et al. (April 2007). "Novel Crohn Disease Locus Identified by Genome-Wide Association Maps to a Gene Desert on 5p13.1 and Modulates Expression of PTGER4". PLOS Genetics. 3 (4): e58. doi:10.1371/journal.pgen.0030058. PMC 1853118. PMID 17447842.
- ↑ Harismendy, Olivier; et al. (10 February 2011). "9p21 DNA Variants Associated with Coronary Artery Disease Impair Interferon-Gamma Signaling Response". Nature. 470 (7333): 264–268. Bibcode:2011Natur.470..264H. doi:10.1038/nature09753. PMC 3079517. PMID 21307941.