| Gómez-López-Hernández syndrome | |
|---|---|
| Other names | Craniosynostosis-alopecia-brain defect syndrome, Craniosynostosis-alopecia-brain defect syndrome |
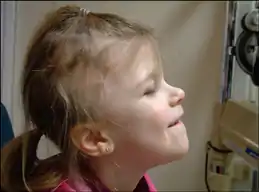 | |
| A young girl showing characteristic bilateral alopecia and angled ears | |
Gómez–López-Hernández syndrome (GLH) or cerebellotrigeminal-dermal dysplasia is a rare neurocutaneous (Phakomatosis) disorder affecting the trigeminal nerve and causing several other neural and physical abnormalities.[1] Gómez–López-Hernández syndrome has been diagnosed in only 34 people.[2] Cases of Gómez–López-Hernández syndrome may be under-reported as other diseases share the characteristics of cerebellar malformation shown in Gómez–López-Hernández syndrome.[1][2] Gómez–López-Hernández syndrome was first characterized in 1979.[3]
Presentation
Physical
Physical characteristics of the syndrome can vary and are not universal. People with Gómez–López-Hernández syndrome often have a short skull (brachycephaly), thin lips, low-set and posterior-angled ears, and scalp alopecia above both ears.[4] This bilateral scalp alopecia is the most consistent physical characteristic of Gómez–López-Hernández syndrome.[5] In addition to the shortness of the skull, it is also misshapen and often flattened on the back.[2] Some people with Gómez–López-Hernández syndrome also have wide-set (hypertelorism) and crossed eyes (strabismus).[4] Scarring or clouding of the cornea occurs in the majority of people with Gómez–López-Hernández syndrome.[6] A short stature is common.[1]
Neurological
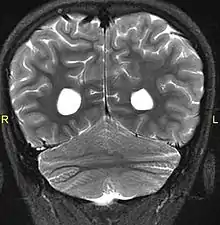
Aside from the physical characteristics of the eyes there is also less sensation in the eyes when stimulated.[4] The eyes also show low motor control (ataxia).[4] Along with ataxia comes a lack of coordination or ability to judge the distance of objects (dysmetria).[6] MRIs show a constant feature of rhombencephalosynapsis–a condition marked by the absence or partial absence of the cerebellar vermis and varying degrees of fusion in the cerebellum in every case of Gómez–López-Hernández syndrome.[4][7][8] Also absent are the trigeminal nerve of the trigeminal cave and the foramen rotundum, causing abnormal sensations on the forehead and the corneas.[6][8] One Gómez–López-Hernández syndrome case in Japan also presents fever-induced seizures.[9] Others may or may not present with non-fever-induced seizures.[4] Malformations of motor centers in the brain cause reduced muscle strength (hypotonia).[8] Eleven of fifteen people in one study showed moderate-to-severe intellectual disability.[6] In cases where it has been noted, head nodding is present.[6] Hydrocephalus and enlargement of the ventricular system is consistently present.[6] A reduced corpus callosum is present in some cases (agenesis of the corpus callosum).[6]
Behavioral
Gómez–López-Hernández syndrome is associated with irritability, anxiety, insomnia, and self-harming behavior.[4] Developmental disabilities often present as intellectual disability with social, occupational, and learning disabilities.[4] Reduced eye sensation may cause self-harm to the eyes; one patient is on record as having put her fingers into her eyes to the point of causing additional corneal damage beyond what is normally characteristic of the syndrome.[4]
Causes
The exact causes of Gómez–López-Hernández syndrome are currently unknown. Mutations of the ACP2 gene have been implicated but not confirmed.[4] One case of siblings — both with Gómez–López-Hernández syndrome — has been observed, showing possible evidence of recessive inheritance.[2] The Brazilian parents of these siblings showed some degree of inbreeding, being first cousins.[2] Five of the 34 people diagnosed with Gómez–López-Hernández syndrome have also come from Brazil.[2] Lack of expression from the WNT1, FGF8, FGF17, OTX2, fgf8, and fgf17 genes have all been implicated as possibly being the cause of cerebellum fusion.[4]
Diagnosis
All cases of Gómez–López-Hernández syndrome present scalp alopecia, varying degrees of low-set ears and most have a flattened skull.[10] Scalp alopecia has been present in all but one case though it can be asymmetrical or, in a single case, only present on one side.[5] All people with Gómez–López-Hernández syndrome also have delayed motor milestones.[10] All people with the syndrome have malformation of the cerebellum.[10] Certain characteristics are often present in those with Gómez–López-Hernández syndrome but are not consistent enough to rule out the syndrome if they are not present. Reduced eye sensation, or trigeminal anesthesia, is present in about three-quarters of people with Gómez–López-Hernández syndrome.[5] Malformations of the skull, rotations of the ears, and abnormalities of the face are features that vary widely and cannot be used alone to diagnose Gómez–López-Hernández syndrome as these characteristics overlap with some other diseases.[5] Gómez–López-Hernández syndrome has been diagnosed as early as 21 weeks with prenatal MRI showing fusion of the cerebellum and later confirmed postnatal with skull and facial abnormalities at six weeks.[11]
Management
Gómez–López-Hernández syndrome is rare and similar to other developmental disabilities. Management is similar to other developmental disabilities as there is no cure for malformations of the brain. Gómez–López-Hernández syndrome has been diagnosed mostly in poorer countries.[4] There have been no documented attempts made to educate children with the syndrome (when intellectual disability is present) to establish a baseline of intellectual ability due to these socioeconomic problems.[4]
Prognosis
The oldest person who has been diagnosed with Gómez–López-Hernández syndrome was 29 years old at the time of his assessment in 2008.[4] Most people with Gómez–López-Hernández syndrome are consistently low weight (3rd-25th percentile) and low stature due to a deficiency of growth hormone.[4] Low mobility is often an issue.[4] The cause of low mobility is thought to be neurological, therefore bone structure is not greatly affected.[4] Seizures may or may not be present and can result in injuries for those who are more mobile.[4] ADHD and bipolar disorder — which are sometimes present — can lead to dangerous behavior or outbursts.[4] While most people with Gómez–López-Hernández syndrome show moderate intellectual disability, one case (age 14) has resulted in normal learning and social skills without intervention.[4]
Epidemiology
Most cases have been diagnosed in Latin America with five of the thirty-four being from Brazil.[2] Two of these Brazilians are related by blood (consanguinity) suggesting the possibility of Mendelian inheritance.[2] There has been one case of a Japanese patient with Gómez–López-Hernández syndrome so far.[9] Two Armenian cases and two from Europe have been identified, signaling that the perceived prevalence in Latin America may be short-lived as better diagnostic techniques and information about this syndrome become more widespread.[5] Recently, a case of Gómez–López-Hernández syndrome was reported from India as well.[12]
Eponym
Gómez–López-Hernández syndrome is named for Manuel Rodríguez Gómez[13] and Alejandro López-Hernández.[14]
References
- 1 2 3 Fernández-Jaén A, Fernández-Mayoralas DM, Calleja-Pérez B, Muñoz-Jareño N, Moreno N (2009). "Gomez-Lopez-Hernandez syndrome: two new cases and review of the literature". Pediatr Neurol. 40 (1): 58–62. doi:10.1016/j.pediatrneurol.2008.10.001. PMID 19068257.
- 1 2 3 4 5 6 7 8 de Mattos VF, Graziadio C, Machado Rosa RF, Lenhardt R, Alves RP, Trevisan P, Paskulin GA, Zen PR (2014). "Gómez-López-Hernández syndrome in a child born to consanguineous parents: new evidence for an autosomal-recessive pattern of inheritance?". Pediatr Neurol. 50 (6): 612–5. doi:10.1016/j.pediatrneurol.2014.01.035. PMID 24690526.
- ↑ Gómez MR (1979). "Cerebellotrigeminal and focal dermal dysplasia: a newly recognized neurocutaneous syndrome". Brain Dev. 1 (4): 253–6. doi:10.1016/s0387-7604(79)80039-x. PMID 95427. S2CID 4698879.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Gomy I, Heck B, Santos AC, Figueiredo MS, Martinelli CE Jr, Nogueira MP, Pina-Neto JM (2008). "Two new Brazilian patients with Gómez-López-Hernández syndrome: reviewing the expanded phenotype with molecular insights". Am J Med Genet A. 146A (5): 649–57. doi:10.1002/ajmg.a.32173. PMID 18247421. S2CID 6105360.
- 1 2 3 4 5 Sukhudyan B, Jaladyan V, Melikyan G, Schlump JU, Boltshauser E, Poretti A (2013). "Gómez-López-Hernández syndrome: reappraisal of the diagnostic criteria" (PDF). Eur. J. Pediatr. 169 (12): 1523–8. doi:10.1007/s00431-010-1259-7. PMID 20652311. S2CID 7101190.
- 1 2 3 4 5 6 7 Poretti A, Bartholdi D, Gobara S, Alber FD, Boltshauser E (2008). "Gomez-Lopez-Hernandez syndrome: an easily missed diagnosis". Eur J Med Genet. 51 (3): 198–208. doi:10.1016/j.ejmg.2008.01.004. PMID 18342593.
- ↑ Abdel-Salam GM, Abdel-Hadi S, Thomas MM, Eid OM, Ali MM, Afifi HH (2014). "Gómez-López-hernández syndrome versus rhombencephalosynapsis spectrum: a rare co-occurrence with bipartite parietal bone". Am J Med Genet A. 164A (2): 480–3. doi:10.1002/ajmg.a.36276. PMID 24311025. S2CID 24479285.
- 1 2 3 Choudhri AF, Patel RM, Wilroy RS, Pivnick EK, Whitehead MT (2015). "GTrigeminal nerve agenesis with absence of foramina rotunda in Gómez-López-Hernández syndrome". Am J Med Genet A. 167A (1): 238–42. doi:10.1002/ajmg.a.36830. PMID 25339626. S2CID 24540712.
- 1 2 Kobayashi Y, Kawashima H, Magara S, Akasaka N, Tohyama J (2015). "Gómez-López-Hernández syndrome in a Japanese patient: a case report". Brain Dev. 37 (3): 356–8. doi:10.1016/j.braindev.2014.05.002. PMID 24856766. S2CID 28604985.
- 1 2 3 Rush ET, Adam MP, Clark RD, Curry C, Hartmann JE, Dobyns WB, Olney AH (2013). "Four new patients with Gomez-Lopez-Hernandez syndrome and proposed diagnostic criteria". Am J Med Genet A. 161A (2): 320–6. doi:10.1002/ajmg.a.35817. PMID 23292994. S2CID 22605993.
- ↑ Tan TY, McGillivray G, Goergen SK, White SM (2005). "GPrenatal magnetic resonance imaging in Gomez-Lopez-Hernandez syndrome and review of the literature". Am J Med Genet A. 138 (4): 369–73. doi:10.1002/ajmg.a.30967. PMID 16158443. S2CID 11532423.
- ↑ Choudhary, Neha; Prabhakar, Anuj; Bhatia, Vikas; Gupta, Parul Chawla (2021-10-19). "Gomez-López-Hernandez syndrome: the triad of cerebello-trigemino-dermal dysplasia". BMJ Case Reports. 14 (10): e246189. doi:10.1136/bcr-2021-246189. ISSN 1757-790X. PMC 8527131. PMID 34667053.
- ↑ Gomez MR (1979). "Cerebellotrigeminal and focal dermal dysplasia: a newly recognized neurocutaneous syndrome". Brain Dev. 1 (4): 253–6. doi:10.1016/s0387-7604(79)80039-x. PMID 95427. S2CID 4698879.
- ↑ López-Hernández, A (2013). "Craniosynostosis, ataxia, trigeminal anaesthesia and parietal alopecia with pons-vermis fusion anomaly (atresia of the fourth ventricle). Report of two cases". Neuropediatrics. 13 (2): 99–102. doi:10.1055/s-2008-1059606. PMID 7133329. S2CID 38892285.