In chemistry, a hypervalent molecule (the phenomenon is sometimes colloquially known as expanded octet) is a molecule that contains one or more main group elements apparently bearing more than eight electrons in their valence shells. Phosphorus pentachloride (PCl5), sulfur hexafluoride (SF6), chlorine trifluoride (ClF3), the chlorite (ClO−2) ion, and the triiodide (I−3) ion are examples of hypervalent molecules.
Definitions and nomenclature
Hypervalent molecules were first formally defined by Jeremy I. Musher in 1969 as molecules having central atoms of group 15–18 in any valence other than the lowest (i.e. 3, 2, 1, 0 for Groups 15, 16, 17, 18 respectively, based on the octet rule).[1]
Several specific classes of hypervalent molecules exist:
- Hypervalent iodine compounds are useful reagents in organic chemistry (e.g. Dess–Martin periodinane)
- Tetra-, penta- and hexavalent phosphorus, silicon, and sulfur compounds (e.g. PCl5, PF5, SF6, sulfuranes and persulfuranes)
- Noble gas compounds (ex. xenon tetrafluoride, XeF4)
- Halogen polyfluorides (ex. chlorine pentafluoride, ClF5)
N-X-L notation
N-X-L nomenclature, introduced collaboratively by the research groups of Martin, Arduengo, and Kochi in 1980,[2] is often used to classify hypervalent compounds of main group elements, where:
- N represents the number of valence electrons
- X is the chemical symbol of the central atom
- L the number of ligands to the central atom
Examples of N-X-L nomenclature include:
History and controversy
The debate over the nature and classification of hypervalent molecules goes back to Gilbert N. Lewis and Irving Langmuir and the debate over the nature of the chemical bond in the 1920s.[3] Lewis maintained the importance of the two-center two-electron (2c-2e) bond in describing hypervalence, thus using expanded octets to account for such molecules. Using the language of orbital hybridization, the bonds of molecules like PF5 and SF6 were said to be constructed from sp3dn orbitals on the central atom. Langmuir, on the other hand, upheld the dominance of the octet rule and preferred the use of ionic bonds to account for hypervalence without violating the rule (e.g. "SF2+
4 2F−" for SF6).
In the late 1920s and 1930s, Sugden argued for the existence of a two-center one-electron (2c-1e) bond and thus rationalized bonding in hypervalent molecules without the need for expanded octets or ionic bond character; this was poorly accepted at the time.[3] In the 1940s and 1950s, Rundle and Pimentel popularized the idea of the three-center four-electron bond, which is essentially the same concept which Sugden attempted to advance decades earlier; the three-center four-electron bond can be alternatively viewed as consisting of two collinear two-center one-electron bonds, with the remaining two nonbonding electrons localized to the ligands.[3]
The attempt to actually prepare hypervalent organic molecules began with Hermann Staudinger and Georg Wittig in the first half of the twentieth century, who sought to challenge the extant valence theory and successfully prepare nitrogen and phosphorus-centered hypervalent molecules.[4] The theoretical basis for hypervalency was not delineated until J.I. Musher's work in 1969.[1]
In 1990, Magnusson published a seminal work definitively excluding the significance of d-orbital hybridization in the bonding of hypervalent compounds of second-row elements. This had long been a point of contention and confusion in describing these molecules using molecular orbital theory. Part of the confusion here originates from the fact that one must include d-functions in the basis sets used to describe these compounds (or else unreasonably high energies and distorted geometries result), and the contribution of the d-function to the molecular wavefunction is large. These facts were historically interpreted to mean that d-orbitals must be involved in bonding. However, Magnusson concludes in his work that d-orbital involvement is not implicated in hypervalency.[5]
Nevertheless, a 2013 study showed that although the Pimentel ionic model best accounts for the bonding of hypervalent species, the energetic contribution of an expanded octet structure is also not null. In this modern valence bond theory study of the bonding of xenon difluoride, it was found that ionic structures account for about 81% of the overall wavefunction, of which 70% arises from ionic structures employing only the p orbital on xenon while 11% arises from ionic structures employing an hybrid on xenon. The contribution of a formally hypervalent structure employing an orbital of sp3d hybridization on xenon accounts for 11% of the wavefunction, with a diradical contribution making up the remaining 8%. The 11% sp3d contribution results in a net stabilization of the molecule by 7.2 kcal (30 kJ) mol−1,[6] a minor but significant fraction of the total energy of the total bond energy (64 kcal (270 kJ) mol−1).[7] Other studies have similarly found minor but non-negligible energetic contributions from expanded octet structures in SF6 (17%) and XeF6 (14%).[8]

Despite the lack of chemical realism, the IUPAC recommends the drawing of expanded octet structures for functional groups like sulfones and phosphoranes, in order to avoid the drawing of a large number of formal charges or partial single bonds.[9]
Hypervalent hydrides
A special type of hypervalent molecules is hypervalent hydrides. Most known hypervalent molecules contain substituents more electronegative than their central atoms.[10][11] Hypervalent hydrides are of special interest because hydrogen is usually less electronegative than the central atom. A number of computational studies have been performed on chalcogen hydrides[11][12][13][14][15][16] and pnictogen hydrides.[17][18][19][20][21] Recently, a new computational study has showed that most hypervalent halogen hydrides XHn can exist. It is suggested that IH3 and IH5 are stable enough to be observable or, possibly, even isolable.[22]
Criticism
Both the term and concept of hypervalency still fall under criticism. In 1984, in response to this general controversy, Paul von Ragué Schleyer proposed the replacement of 'hypervalency' with use of the term hypercoordination because this term does not imply any mode of chemical bonding and the question could thus be avoided altogether.[3]
The concept itself has been criticized by Ronald Gillespie who, based on an analysis of electron localization functions, wrote in 2002 that "as there is no fundamental difference between the bonds in hypervalent and non-hypervalent (Lewis octet) molecules there is no reason to continue to use the term hypervalent."[23]
For hypercoordinated molecules with electronegative ligands such as PF5, it has been demonstrated that the ligands can pull away enough electron density from the central atom so that its net content is again 8 electrons or fewer. Consistent with this alternative view is the finding that hypercoordinated molecules based on fluorine ligands, for example PF5 do not have hydride counterparts, e.g. phosphorane (PH5) which is unknown.
The ionic model holds up well in thermochemical calculations. It predicts favorable exothermic formation of PF+
4F−
from phosphorus trifluoride PF3 and fluorine F2 whereas a similar reaction forming PH+
4H−
is not favorable.[24]
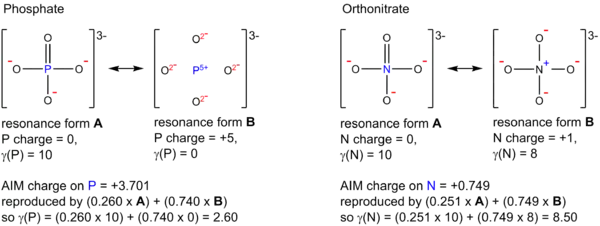
Alternative definition
Durrant has proposed an alternative definition of hypervalency, based on the analysis of atomic charge maps obtained from atoms in molecules theory.[25] This approach defines a parameter called the valence electron equivalent, γ, as “the formal shared electron count at a given atom, obtained by any combination of valid ionic and covalent resonance forms that reproduces the observed charge distribution”. For any particular atom X, if the value of γ(X) is greater than 8, that atom is hypervalent. Using this alternative definition, many species such as PCl5, SO2−
4, and XeF4, that are hypervalent by Musher's definition, are reclassified as hypercoordinate but not hypervalent, due to strongly ionic bonding that draws electrons away from the central atom. On the other hand, some compounds that are normally written with ionic bonds in order to conform to the octet rule, such as ozone O3, nitrous oxide NNO, and trimethylamine N-oxide (CH
3)
3NO, are found to be genuinely hypervalent. Examples of γ calculations for phosphate PO3−
4 (γ(P) = 2.6, non-hypervalent) and orthonitrate NO3−
4 (γ(N) = 8.5, hypervalent) are shown below.
Bonding in hypervalent molecules
Early considerations of the geometry of hypervalent molecules returned familiar arrangements that were well explained by the VSEPR model for atomic bonding. Accordingly, AB5 and AB6 type molecules would possess a trigonal bi-pyramidal and octahedral geometry, respectively. However, in order to account for the observed bond angles, bond lengths and apparent violation of the Lewis octet rule, several alternative models have been proposed.
In the 1950s an expanded valence shell treatment of hypervalent bonding was adduced to explain the molecular architecture, where the central atom of penta- and hexacoordinated molecules would utilize d AOs in addition to s and p AOs. However, advances in the study of ab initio calculations have revealed that the contribution of d-orbitals to hypervalent bonding is too small to describe the bonding properties, and this description is now regarded as much less important.[5] It was shown that in the case of hexacoordinated SF6, d-orbitals are not involved in S-F bond formation, but charge transfer between the sulfur and fluorine atoms and the apposite resonance structures were able to account for the hypervalency (See below).
Additional modifications to the octet rule have been attempted to involve ionic characteristics in hypervalent bonding. As one of these modifications, in 1951, the concept of the 3-center 4-electron (3c-4e) bond, which described hypervalent bonding with a qualitative molecular orbital, was proposed. The 3c-4e bond is described as three molecular orbitals given by the combination of a p atomic orbital on the central atom and an atomic orbital from each of the two ligands on opposite sides of the central atom. Only one of the two pairs of electrons is occupying a molecular orbital that involves bonding to the central atom, the second pair being non-bonding and occupying a molecular orbital composed of only atomic orbitals from the two ligands. This model in which the octet rule is preserved was also advocated by Musher.[3]

Molecular orbital theory
A complete description of hypervalent molecules arises from consideration of molecular orbital theory through quantum mechanical methods. An LCAO in, for example, sulfur hexafluoride, taking a basis set of the one sulfur 3s-orbital, the three sulfur 3p-orbitals, and six octahedral geometry symmetry-adapted linear combinations (SALCs) of fluorine orbitals, a total of ten molecular orbitals are obtained (four fully occupied bonding MOs of the lowest energy, two fully occupied intermediate energy non-bonding MOs and four vacant antibonding MOs with the highest energy) providing room for all 12 valence electrons. This is a stable configuration only for SX6 molecules containing electronegative ligand atoms like fluorine, which explains why SH6 is not a stable molecule. In the bonding model, the two non-bonding MOs (1eg) are localized equally on all six fluorine atoms.
Valence bond theory
For hypervalent compounds in which the ligands are more electronegative than the central, hypervalent atom, resonance structures can be drawn with no more than four covalent electron pair bonds and completed with ionic bonds to obey the octet rule. For example, in phosphorus pentafluoride (PF5), 5 resonance structures can be generated each with four covalent bonds and one ionic bond with greater weight in the structures placing ionic character in the axial bonds, thus satisfying the octet rule and explaining both the observed trigonal bipyramidal molecular geometry and the fact that the axial bond length (158 pm) is longer than the equatorial (154 pm).[26]
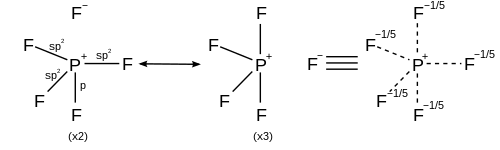
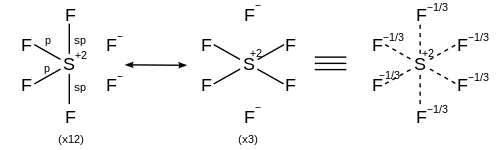
For a hexacoordinate molecule such as sulfur hexafluoride, each of the six bonds is the same length. The rationalization described above can be applied to generate 15 resonance structures each with four covalent bonds and two ionic bonds, such that the ionic character is distributed equally across each of the sulfur-fluorine bonds.
Spin-coupled valence bond theory has been applied to diazomethane and the resulting orbital analysis was interpreted in terms of a chemical structure in which the central nitrogen has five covalent bonds;
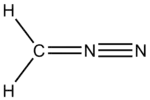
This led the authors to the interesting conclusion that "Contrary to what we were all taught as undergraduates, the nitrogen atom does indeed form five covalent linkages and the availability or otherwise of d-orbitals has nothing to do with this state of affairs."[27]
Structure, reactivity, and kinetics
Structure
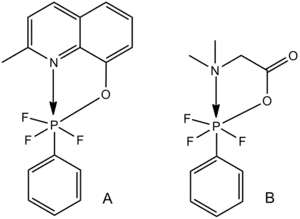
Hexacoordinated phosphorus
Hexacoordinate phosphorus molecules involving nitrogen, oxygen, or sulfur ligands provide examples of Lewis acid-Lewis base hexacoordination.[28] For the two similar complexes shown below, the length of the C–P bond increases with decreasing length of the N–P bond; the strength of the C–P bond decreases with increasing strength of the N–P Lewis acid–Lewis base interaction.
Pentacoordinated silicon
This trend is also generally true of pentacoordinated main-group elements with one or more lone-pair-containing ligand, including the oxygen-pentacoordinated silicon examples shown below.
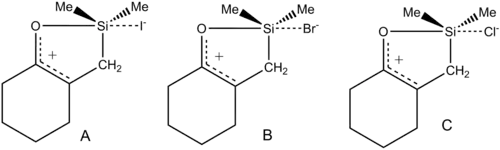
The Si-halogen bonds range from close to the expected van der Waals value in A (a weak bond) almost to the expected covalent single bond value in C (a strong bond).[28]
Reactivity
Silicon
| Chlorosilane | Nucleophile | kobs (M−2s−1) at 20 °C in anisole |
|---|---|---|
| Ph3SiCl | HMPT | 1200 |
| Ph3SiCl | DMSO | 50 |
| Ph3SiCl | DMF | 6 |
| MePh2SiCl | HMPT | 2000 |
| MePh2SiCl | DMSO | 360 |
| MePh2SiCl | DMF | 80 |
| Me(1-Np)PhSiCl | HMPT | 3500 |
| Me(1-Np)PhSiCl | DMSO | 180 |
| Me(1-Np)PhSiCl | DMF | 40 |
| (1-Np)Ph(vinyl)SiCl | HMPT | 2200 |
| (1-Np)Ph(vinyl)SiCl | DMSO | 90 |
| (1-Np)(m-CF3Ph)HSiCl | DMSO | 1800 |
| (1-Np)(m-CF3Ph)HSiCl | DMF | 300 |
Corriu and coworkers performed early work characterizing reactions thought to proceed through a hypervalent transition state.[29] Measurements of the reaction rates of hydrolysis of tetravalent chlorosilanes incubated with catalytic amounts of water returned a rate that is first order in chlorosilane and second order in water. This indicated that two water molecules interacted with the silane during hydrolysis and from this a binucleophilic reaction mechanism was proposed. Corriu and coworkers then measured the rates of hydrolysis in the presence of nucleophilic catalyst HMPT, DMSO or DMF. It was shown that the rate of hydrolysis was again first order in chlorosilane, first order in catalyst and now first order in water. Appropriately, the rates of hydrolysis also exhibited a dependence on the magnitude of charge on the oxygen of the nucleophile.
Taken together this led the group to propose a reaction mechanism in which there is a pre-rate determining nucleophilic attack of the tetracoordinated silane by the nucleophile (or water) in which a hypervalent pentacoordinated silane is formed. This is followed by a nucleophilic attack of the intermediate by water in a rate determining step leading to hexacoordinated species that quickly decomposes giving the hydroxysilane.
Silane hydrolysis was further investigated by Holmes and coworkers [30] in which tetracoordinated Mes
2SiF
2 (Mes = mesityl) and pentacoordinated Mes
2SiF−
3 were reacted with two equivalents of water. Following twenty-four hours, almost no hydrolysis of the tetracoordinated silane was observed, while the pentacoordinated silane was completely hydrolyzed after fifteen minutes. Additionally, X-ray diffraction data collected for the tetraethylammonium salts of the fluorosilanes showed the formation of hydrogen bisilonate lattice supporting a hexacoordinated intermediate from which HF−
2 is quickly displaced leading to the hydroxylated product. This reaction and crystallographic data support the mechanism proposed by Corriu et al..

The apparent increased reactivity of hypervalent molecules, contrasted with tetravalent analogues, has also been observed for Grignard reactions. The Corriu group measured[31] Grignard reaction half-times by NMR for related 18-crown-6 potassium salts of a variety of tetra- and pentacoordinated fluorosilanes in the presence of catalytic amounts of nucleophile.
Though the half reaction method is imprecise, the magnitudinal differences in reactions rates allowed for a proposed reaction scheme wherein, a pre-rate determining attack of the tetravalent silane by the nucleophile results in an equilibrium between the neutral tetracoordinated species and the anionic pentavalent compound. This is followed by nucleophilic coordination by two Grignard reagents as normally seen, forming a hexacoordinated transition state and yielding the expected product.
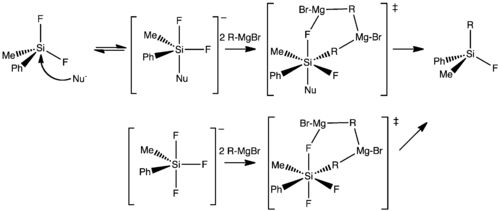
The mechanistic implications of this are extended to a hexacoordinated silicon species that is thought to be active as a transition state in some reactions. The reaction of allyl- or crotyl-trifluorosilanes with aldehydes and ketones only precedes with fluoride activation to give a pentacoordinated silicon. This intermediate then acts as a Lewis acid to coordinate with the carbonyl oxygen atom. The further weakening of the silicon–carbon bond as the silicon becomes hexacoordinate helps drive this reaction.[32]
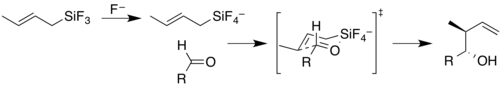
Phosphorus
Similar reactivity has also been observed for other hypervalent structures such as the miscellany of phosphorus compounds, for which hexacoordinated transition states have been proposed. Hydrolysis of phosphoranes and oxyphosphoranes have been studied [33] and shown to be second order in water. Bel'skii et al.. have proposed a prerate determining nucleophilic attack by water resulting in an equilibrium between the penta- and hexacoordinated phosphorus species, which is followed by a proton transfer involving the second water molecule in a rate determining ring-opening step, leading to the hydroxylated product.

Alcoholysis of pentacoordinated phosphorus compounds, such as trimethoxyphospholene with benzyl alcohol, have also been postulated to occur through a similar octahedral transition state, as in hydrolysis, however without ring opening.[34]

It can be understood from these experiments that the increased reactivity observed for hypervalent molecules, contrasted with analogous nonhypervalent compounds, can be attributed to the congruence of these species to the hypercoordinated activated states normally formed during the course of the reaction.
Ab initio calculations
The enhanced reactivity at pentacoordinated silicon is not fully understood. Corriu and coworkers suggested that greater electropositive character at the pentavalent silicon atom may be responsible for its increased reactivity.[35] Preliminary ab initio calculations supported this hypothesis to some degree, but used a small basis set.[36]
A software program for ab initio calculations, Gaussian 86, was used by Dieters and coworkers to compare tetracoordinated silicon and phosphorus to their pentacoordinate analogues. This ab initio approach is used as a supplement to determine why reactivity improves in nucleophilic reactions with pentacoordinated compounds. For silicon, the 6-31+G* basis set was used because of its pentacoordinated anionic character and for phosphorus, the 6-31G* basis set was used.[36]
Pentacoordinated compounds should theoretically be less electrophilic than tetracoordinated analogues due to steric hindrance and greater electron density from the ligands, yet experimentally show greater reactivity with nucleophiles than their tetracoordinated analogues. Advanced ab initio calculations were performed on series of tetracoordinated and pentacoordinated species to further understand this reactivity phenomenon. Each series varied by degree of fluorination. Bond lengths and charge densities are shown as functions of how many hydride ligands are on the central atoms. For every new hydride, there is one less fluoride.[36]
For silicon and phosphorus bond lengths, charge densities, and Mulliken bond overlap, populations were calculated for tetra and pentacoordinated species by this ab initio approach.[36] Addition of a fluoride ion to tetracoordinated silicon shows an overall average increase of 0.1 electron charge, which is considered insignificant. In general, bond lengths in trigonal bipyramidal pentacoordinate species are longer than those in tetracoordinate analogues. Si-F bonds and Si-H bonds both increase in length upon pentacoordination and related effects are seen in phosphorus species, but to a lesser degree. The reason for the greater magnitude in bond length change for silicon species over phosphorus species is the increased effective nuclear charge at phosphorus. Therefore, silicon is concluded to be more loosely bound to its ligands.

In addition Dieters and coworkers [36] show an inverse correlation between bond length and bond overlap for all series. Pentacoordinated species are concluded to be more reactive because of their looser bonds as trigonal-bipyramidal structures.
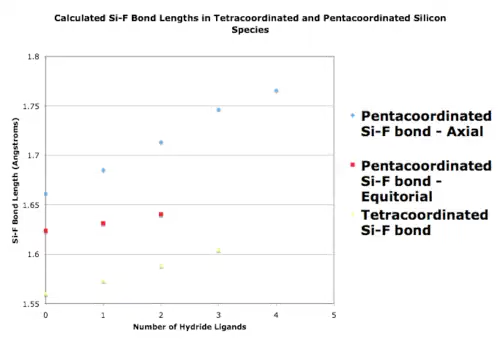

By calculating the energies for the addition and removal of a fluoride ion in various silicon and phosphorus species, several trends were found. In particular, the tetracoordinated species have much higher energy requirements for ligand removal than do pentacoordinated species. Further, silicon species have lower energy requirements for ligand removal than do phosphorus species, which is an indication of weaker bonds in silicon.
See also
References
- 1 2 Musher, J.I. (1969). "The Chemistry of Hypervalent Molecules". Angew. Chem. Int. Ed. 8: 54–68. doi:10.1002/anie.196900541.
- ↑ Perkins, C. W.; Martin, J. C.; Arduengo, A. J.; Lau, W.; Alegria, A; Kochi, J. K. (1980). "An Electrically Neutral σ-Sulfuranyl Radical from the Homolysis of a Perester with Neighboring Sulfenyl Sulfur: 9-S-3 species". J. Am. Chem. Soc. 102 (26): 7753–7759. doi:10.1021/ja00546a019.
- 1 2 3 4 5 Jensen, W. (2006). "The Origin of the Term "Hypervalent"". J. Chem. Educ. 83 (12): 1751. Bibcode:2006JChEd..83.1751J. doi:10.1021/ed083p1751. | Link
- ↑ Kin-ya Akiba (1999). Chemistry of Hypervalent Compounds. New York: Wiley VCH. ISBN 978-0-471-24019-8.
- 1 2 Magnusson, E. (1990). "Hypercoordinate molecules of second-row elements: d functions or d orbitals?". J. Am. Chem. Soc. 112 (22): 7940–7951. doi:10.1021/ja00178a014.
- ↑ Braïda, Benoît; Hiberty, Philippe C. (2013-04-07). "The essential role of charge-shift bonding in hypervalent prototype XeF2" (PDF). Nature Chemistry. 5 (5): 417–422. Bibcode:2013NatCh...5..417B. doi:10.1038/nchem.1619. ISSN 1755-4330. PMID 23609093.
- ↑ H., Cockett, A. (2013). The Chemistry of the Monatomic Gases : Pergamon Texts in Inorganic Chemistry. Smith, K. C., Bartlett, Neil. Saint Louis: Elsevier Science. ISBN 9781483157368. OCLC 953379200.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ Lein, Matthias; Frenking, Gernot (2005-01-01). "The nature of the chemical bond in the light of an energy decomposition analysis". Theory and Applications of Computational Chemistry: 291–372. doi:10.1016/B978-044451719-7/50056-1. ISBN 9780444517197.
- ↑ Brecher, Jonathan (2008). "Graphical representation standards for chemical structure diagrams (IUPAC Recommendations 2008)". Pure and Applied Chemistry. 80 (2): 277–410. doi:10.1351/pac200880020277. ISSN 0033-4545.
- ↑ Reed, Alan E.; Schleyer, Paul v. R. (November 1988). "The anomeric effect with central atoms other than carbon. 2. Strong interactions between nonbonded substituents in mono- and polyfluorinated first- and second-row amines, FnAHmNH2". Inorganic Chemistry. 27 (22): 3969–3987. doi:10.1021/ic00295a018. ISSN 0020-1669.
- 1 2 Pu, Zhifeng; Li, Qian-shu; Xie, Yaoming; Schaefer, Henry F. (October 2009). "Hypervalent molecules, sulfuranes, and persulfuranes: review and studies related to the recent synthesis of the first persulfurane with all substituents carbon-linked". Theoretical Chemistry Accounts. 124 (3–4): 151–159. doi:10.1007/s00214-009-0621-1. ISSN 1432-881X. S2CID 96331962.
- ↑ Yoshioka, Yasunori; Goddard, John D.; Schaefer, Henry F. (February 1981). "Analytic configuration interaction gradient studies of SH 4 , sulfurane". The Journal of Chemical Physics. 74 (3): 1855–1863. Bibcode:1981JChPh..74.1855Y. doi:10.1063/1.441275. ISSN 0021-9606.
- ↑ Moc, Jerzy; Dorigo, Andrea E.; Morokuma, Keiji (March 1993). "Transition structures for H2 elimination from XH4 hypervalent species (X = S, Se and Te). Ab initio MO study". Chemical Physics Letters. 204 (1–2): 65–72. Bibcode:1993CPL...204...65M. doi:10.1016/0009-2614(93)85606-O.
- ↑ Wittkopp, Alexander; Prall, Matthias; Schreiner, Peter R.; Schaefer III, Henry F. (2000). "Is SH4, the simplest 10-S-4 sulfurane, observable?". Physical Chemistry Chemical Physics. 2 (10): 2239–2244. Bibcode:2000PCCP....2.2239W. doi:10.1039/b000597p.
- ↑ Schwenzer, Gretchen M.; Schaefer, Henry F. III (March 1975). "Hypervalent molecules sulfurane (SH4) and persulfurane (SH6)". Journal of the American Chemical Society. 97 (6): 1393–1397. doi:10.1021/ja00839a019. ISSN 0002-7863. S2CID 93412551.
- ↑ Hinze, Juergen; Friedrich, Oliver; Sundermann, Andreas (February 1999). "A study of some unusual hydrides: BeH2, BeH+6 and SH6". Molecular Physics. 96 (4): 711–718. Bibcode:1999MolPh..96..711H. doi:10.1080/00268979909483007. ISSN 0026-8976.
- ↑ Rauk, Arvi; Allen, Leland C.; Mislow, Kurt (May 1972). "Electronic structure of PH5 and intramolecular ligand exchange in phosphoranes. Model studies". Journal of the American Chemical Society. 94 (9): 3035–3040. doi:10.1021/ja00764a026. ISSN 0002-7863.
- ↑ Kutzelnigg, Werner; Wasilewski, Jan (February 1982). "Theoretical study of the reaction PH5 → PH3 + H2". Journal of the American Chemical Society. 104 (4): 953–960. doi:10.1021/ja00368a005. ISSN 0002-7863.
- ↑ Wasada, H.; Hirao, K. (January 1992). "Theoretical study of the reactions of pentacoordinated trigonal bipyramidal phosphorus compounds: PH5, PF5, PF4H, PF3H2, PF4CH3, PF3(CH3)2, P(O2C2H4)H3, P(OC3H6)H3, and PO5H4-". Journal of the American Chemical Society. 114 (1): 16–27. doi:10.1021/ja00027a002. ISSN 0002-7863.
- ↑ Kolandaivel, P.; Kumaresan, R. (August 1995). "The reaction path of PH5 → PH3 + H2 using an SCF study". Journal of Molecular Structure: THEOCHEM. 337 (3): 225–229. doi:10.1016/0166-1280(94)04103-Y.
- ↑ Moc, Jerzy; Morokuma, Keiji (November 1995). "AB Initio Molecular Orbital Study on the Periodic Trends in Structures and Energies of Hypervalent Compounds: Five-Coordinated XH5 Species Containing a Group 5 Central Atom (X = P, As, Sb, and Bi)". Journal of the American Chemical Society. 117 (47): 11790–11797. doi:10.1021/ja00152a022. ISSN 0002-7863.
- ↑ Sikalov, Alexander A. (12 December 2019). "Hypervalent halogen hydrides HalHn (Hal = Cl, Br, I; n = 3, 5, 7): DFT and ab initio stability prediction". Theoretical Chemistry Accounts. 139 (1): 8. doi:10.1007/s00214-019-2524-0. ISSN 1432-2234. S2CID 209331619.
- ↑ Gillespie, R (2002). "The octet rule and hypervalence: Two misunderstood concepts". Coordination Chemistry Reviews. 233–234: 53–62. doi:10.1016/S0010-8545(02)00102-9.
- ↑ Predicting the Stability of Hypervalent Molecules Mitchell, Tracy A.; Finocchio, Debbie; Kua, Jeremy. J. Chem. Educ. 2007, 84, 629. Link
- ↑ Durrant, M. C. (2015). "A quantitative definition of hypervalency" (PDF). Chemical Science. 6 (11): 6614–6623. doi:10.1039/C5SC02076J. PMC 6054109. PMID 30090275.
- ↑ Curnow, Owen J. (1998). "A Simple Qualitative Molecular-Orbital/Valence-Bond Description of the Bonding in Main Group "Hypervalent" Molecules". Journal of Chemical Education. 75 (7): 910–915. Bibcode:1998JChEd..75..910C. doi:10.1021/ed075p910.
- ↑ Gerratt, Joe (1997). "Modern valence bond theory". Chemical Society Reviews. 26 (2): 87–100. doi:10.1039/CS9972600087.
- 1 2 3 4 Holmes, R.R. (1996). "Comparison of Phosphorus and Silicon: Hypervalency, Stereochemistry, and Reactivity". Chem. Rev. 96 (3): 927–950. doi:10.1021/cr950243n. PMID 11848776.
- 1 2 Corriu, RJP; Dabosi, G.; Martineau, M. (1978). "Mécanisme de l'hydrolyse des chlorosilanes, catalysée par un nucléophile: étude cinétique et mise en evidence d'un intermediaire hexacoordonné". J. Organomet. Chem. 150: 27–38. doi:10.1016/S0022-328X(00)85545-X.
- ↑ Johnson, SE; Deiters, JA; Day, RO; Holmes, RR (1989). "Pentacoordinated molecules. 76. Novel hydrolysis pathways of dimesityldifluorosilane via an anionic five-coordinated silicate and a hydrogen-bonded bisilonate. Model intermediates in the sol-gel process". J. Am. Chem. Soc. 111 (9): 3250. doi:10.1021/ja00191a023.
- ↑ Corriu, RJP; Guerin, Christian.; Henner, Bernard J. L.; Wong Chi Man, W. W. C. (1988). "Pentacoordinated silicon anions: reactivity toward strong nucleophiles". Organometallics. 7: 237–8. doi:10.1021/om00091a038.
- ↑ Kira, M; Kobayashi, M.; Sakurai, H. (1987). "Regiospecific and highly stereoselective allylation of aldehydes with allyltrifluorosilane activated by fluoride ions". Tetrahedron Letters. 28 (35): 4081–4084. doi:10.1016/S0040-4039(01)83867-3.
- ↑ Bel'Skii, VE (1979). J. Gen. Chem. USSR. 49: 298.
{{cite journal}}: Missing or empty|title=(help) - ↑ Ramirez, F; Tasaka, K.; Desai, N. B.; Smith, Curtis Page. (1968). "Nucleophilic substitutions at pentavalent phosphorus. Reaction of 2,2,2-trialkoxy-2,2-dihydro-1,3,2-dioxaphospholenes with alcohols". J. Am. Chem. Soc. 90 (3): 751. doi:10.1021/ja01005a035.
- ↑ Brefort, Jean Louis; Corriu, Robert J. P.; Guerin, Christian; Henner, Bernard J. L.; Wong Chi Man, Wong Wee Choy (1990). "Pentacoordinated silicon anions: Synthesis and reactivity". Organometallics. 9 (7): 2080. doi:10.1021/om00157a016.
- 1 2 3 4 5 Dieters, J. A.; Holmes, R. R. (1990). "Enhanced Reactivity of Pentacoordinated Silicon Species. An ab Initio Approach". J. Am. Chem. Soc. 112 (20): 7197–7202. doi:10.1021/ja00176a018.
External links
 Media related to Hypervalent molecules at Wikimedia Commons
Media related to Hypervalent molecules at Wikimedia Commons


