INK4 is a family of cyclin-dependent kinase inhibitors (CKIs). The members of this family (p16INK4a, p15INK4b, p18INK4c, p19INK4d) are inhibitors of CDK4 (hence their name INhibitors of CDK4), and of CDK6. The other family of CKIs, CIP/KIP proteins are capable of inhibiting all CDKs. Enforced expression of INK4 proteins can lead to G1 arrest by promoting redistribution of Cip/Kip proteins and blocking cyclin E-CDK2 activity. In cycling cells, there is a resassortment of Cip/Kip proteins between CDK4/5 and CDK2 as cells progress through G1.[1] Their function, inhibiting CDK4/6, is to block progression of the cell cycle beyond the G1 restriction point.[2] In addition, INK4 proteins play roles in cellular senescence, apoptosis and DNA repair.[3]
INK4 proteins are tumor suppressors and loss-of-function mutations lead to carcinogenesis.[4]
INK4 proteins are highly similar in terms of structure and function, with up to 85% amino acid similarity.[1] They contain multiple ankyrin repeats.[3]
Genes
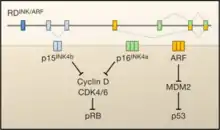
The INK4a/ARF/INK4b locus encodes three genes (p15INK4b, ARF, and p16INK4a) in a 35-kilobase stretch of the human genome. P15INK4b has a different reading frame that is physically separated from p16INK4a and ARF. P16INK4a and ARF have different first exons that are spliced to the same second and third exon. While those second and third exons are shared by p16INK4a and ARF, the proteins are encoded in different reading frames meaning that p16INK4a and ARF are not isoforms, nor do they share any amino acid homology.[1]
Evolution
Polymorphisms of the p15INK4b/p16INK4a homolog were found to segregate with melanoma susceptibility in Xiphophorus indicating that INK4 proteins have been involved with tumor suppression for over 350 million years. Furthermore, the older INK4-based system has been further bolstered by the evolution of the recent addition of the ARF-based anti-cancer response.[1]
Function
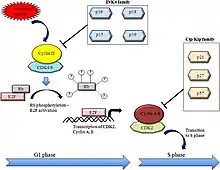
INK4 proteins are cell-cycle inhibitors. When they bind to CDK4 and CDK6, they induce an allosteric change that leads to the formation of CDK-INK4 complexes rather than CDK-cyclin complexes. This leads to an inhibition of retinoblastoma (Rb) phosphorylation downstream. Therefore, the expression of p15INK4b or p16INK4A keeps the Rb-family proteins hypophosphorylated. This allows the hypophosphorylated Rb to repress transcription of S-phase genes causing cell cycle arrest in the G1 phase.[5]
Subsets
P16INK4a
P16 is formed from four ankyrin repeat (AR) motifs that exhibit a helix-turn-helix conformation except that the first helix in the second AR consists of four residues.[6] P16 regulation involves epigenetic control and multiple transcription factors. PRC1, PRC2, YY1, and Id1 play a role in the suppression of p16INK4A expression and transcription factors CTCF, Sp1, and ETs activate p16INK4A transcription.[7] In knockout experiments, it was found that mice lacking just p16INK4a were more prone to spontaneous cancers. Mice lacking both p16INK4a and ARF were found to be even more tumor prone than the mice lacking just p16INK4a.[1]
P15INK4b
P15 is also formed from four ankyrin repeat (AR) motifs. Expression of P15INK4b is induced by TGF-b indicating its role as a potential downstream effector of TGF-b mediated growth arrest.[8]
P18INK4c
P18INK4c has been shown to play an important role in modulating TCR-mediated T cell proliferation. The loss of p18INK4c in T cells reduced the requirement of CD28 costimulation for efficient T cell proliferation. Other INK4 family members did not affect this process. Furthermore, it was shown that p18INK4c is preferentially inhibitory to CDK6, but not CDK4 activity in activated T cells that suggest p18INK4c may set an inhibitory threshold in resting T cells.[9]
Clinical significance
Role in cancer
Cells containing oncogenic mutations in-vivo often responded by activating the INK4A/ARF/INK4B locus that encodes the INK4 tumor suppressor proteins. The unusual genomic arrangement of the INK4a/ARF/INK4b locus functions as a weakness in our anti-cancer defenses. This is due to the fact that three crucial regulators of the RB and p53 (regulated by ARF) are vulnerable to one single, small deletion. This observation yields two possible opposing conclusions: Either tumor formation does not provide any evolutionary selection pressure because the overlapping INK4a/ARF/INK4b is not selected against or tumorigenesis provides such a strong pressure, that an entire group of genes has been selected for at the INK4a/ARF/INK4b locus to prevent cancer. The response of the INK4a/ARF/INK4b locus efficiently prevents cancers that could occur to the constant oncogenic mutations that occur in long-lived mammals.
When the INK4a/ARF/INK4b locus was overexpressed, the mice demonstrated a 3-fold reduction in the incidence of spontaneous cancers. This evidence further indicated that the INK4a/ARF/INK4b locus in mice plays a role in tumor suppression.[1]
Role in aging
The INK4 family has been implicated in the aging process. The expression of p16INK4a increases with aging in many tissues of rodents and humans.[1] It was also shown that INK4a/ARF deficient animals increase an age-related decline in T-cell responsiveness to CD3 and CD28, which is a hallmark of aging. Furthermore, neural stem cells from Bmi-1- deficient animals demonstrate increased INK4a/ARF expression and impaired regenerative potential. The phenotype; however, can be rescued by p16INK4a deficiency implying that while p16INK4a can potentially be used as a biomarker of physiologic, rather than chronologic age, it is also an effector of aging. The mechanism by which it does this is by limiting the self-renewal capacity of disparate tissues such as lymphoid organs, bone marrow, and the brain.[10]
Regulation of INK4 expression
Initially, it was thought that each INK4 family member was structurally redundant and equally potent. It was later found; however, that INK4 family members are differentially expressed during mouse development. The diversity in expression pattern indicates that the INK4 gene family may have cell lineage-specific or tissue-specific functions.[11] Evidence has shown that INK4a/ARF expression increase at an early stage of tumorigenesis, but the precise stimuli relevant to cancer that induces the expression of the locus is unknown. Expression of p15INK4b does not correlate with p16INK4a in many normal rodent tissues. Induction and repression of p15INK4b; however, has been noted in response to a few signaling events such as RAS activation, that also induce INK4/ARF expression. RAS activation might lead to increased INK4/ARF expression potentially through ERK-mediated activation of Ets1/2 to induce p16INK4. A few repressors of INK4a/ARF/INK4b expression have been identified as well. T box proteins and the polycomb group have been shown to repress p16INK4a, p15INK4b, and ARF.[1]
References
- 1 2 3 4 5 6 7 8 9 10 Kim WY, Sharpless NE (October 2006). "The regulation of INK4/ARF in cancer and aging". Cell. 127 (2): 265–75. doi:10.1016/j.cell.2006.10.003. PMID 17055429. S2CID 18280031.
- ↑ Ortega S, Malumbres M, Barbacid M (March 2002). "Cyclin D-dependent kinases, INK4 inhibitors and cancer". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1602 (1): 73–87. doi:10.1016/S0304-419X(02)00037-9. PMID 11960696.
- 1 2 Cánepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF (July 2007). "INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions". IUBMB Life. 59 (7): 419–26. doi:10.1080/15216540701488358. hdl:11336/30965. PMID 17654117. S2CID 9339088.
- ↑ Roussel MF (September 1999). "The INK4 family of cell cycle inhibitors in cancer". Oncogene. 18 (38): 5311–7. doi:10.1038/sj.onc.1202998. PMID 10498883.
- ↑ Sherr CJ (July 2000). "The Pezcoller lecture: cancer cell cycles revisited". Cancer Research. 60 (14): 3689–95. PMID 10919634.
- ↑ Li J, Poi MJ, Tsai MD (June 2011). "Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer". Biochemistry. 50 (25): 5566–82. doi:10.1021/bi200642e. PMC 3127263. PMID 21619050.
- ↑ Rayess H, Wang MB, Srivatsan ES (April 2012). "Cellular senescence and tumor suppressor gene p16". International Journal of Cancer. 130 (8): 1715–25. doi:10.1002/ijc.27316. PMC 3288293. PMID 22025288.
- ↑ Fares J, Wolff L, Bies J (January 2011). Huret JL, Ohgami RS, Dal Cin P, Rivas JM, Dessen P (eds.). "CDKN2B (cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4))". Atlas of genetics and cytogenetics in oncology and haematology. Retrieved 2018-12-16.
- ↑ Kovalev GI, Franklin DS, Coffield VM, Xiong Y, Su L (September 2001). "An important role of CDK inhibitor p18(INK4c) in modulating antigen receptor-mediated T cell proliferation". Journal of Immunology. 167 (6): 3285–92. doi:10.4049/jimmunol.167.6.3285. PMC 4435948. PMID 11544316.
- ↑ Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE (November 2004). "Ink4a/Arf expression is a biomarker of aging". The Journal of Clinical Investigation. 114 (9): 1299–307. doi:10.1172/JCI22475. PMC 524230. PMID 15520862.
- ↑ Cánepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF (July 2007). "INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions". IUBMB Life. 59 (7): 419–26. doi:10.1080/15216540701488358. hdl:11336/30965. PMID 17654117. S2CID 9339088.