Ribosomally synthesized and post-translationally modified peptides (RiPPs), also known as ribosomal natural products, are a diverse class of natural products of ribosomal origin.[1] Consisting of more than 20 sub-classes, RiPPs are produced by a variety of organisms, including prokaryotes, eukaryotes, and archaea, and they possess a wide range of biological functions.
As a consequence of the falling cost of genome sequencing and the accompanying rise in available genomic data, scientific interest in RiPPs has increased in the last few decades. Because the chemical structures of RiPPs are more closely predictable from genomic data than are other natural products (e.g. alkaloids, terpenoids), their presence in sequenced organisms can, in theory, be identified rapidly. This makes RiPPs an attractive target of modern natural product discovery efforts.
Definition
RiPPs consist of any peptides (i.e. molecular weight below 10 kDa) that are ribosomally-produced and undergo some degree of enzymatic post-translational modification. This combination of peptide translation and modification is referred to as "post-ribosomal peptide synthesis" (PRPS) in analogy with nonribosomal peptide synthesis (NRPS).
Historically, the current sub-classes of RiPPs were studied individually, and common practices in nomenclature varied accordingly in the literature. More recently, with the advent of broad genome sequencing, it has been realized that these natural products share a common biosynthetic origin. In 2013, a set of uniform nomenclature guidelines were agreed upon and published by a large group of researchers in the field.[1] Prior to this report, RiPPs were referred to by a variety of designations, including post-ribosomal peptides, ribosomal natural products, and ribosomal peptides.
The acronym "RiPP" stands for "ribosomally synthesized and post-translationally modified peptide".
Prevalence and applications
RiPPs constitute one of the major superfamilies of natural products, like alkaloids, terpenoids, and nonribosomal peptides, although they tend to be large, with molecular weights commonly in excess of 1000 Da.[1] The advent of next-generation sequencing methods has made genome mining of RiPPs a common strategy.[2] In part due to their increased discovery and hypothesized ease of engineering, the use of RiPPs as drugs is increasing. Although they are ribosomal peptides in origin, RiPPs are typically categorized as small molecules rather than biologics due to their chemical properties, such as moderate molecular weight and relatively high hydrophobicity.
The uses and biological activities of RiPPs are diverse.
RiPPs in commercial use include nisin, a food preservative, thiostrepton, a veterinary topical antibiotic, and nosiheptide and duramycin, which are animal feed additives. Phalloidin functionalized with a fluorophore is used in microscopy as a stain due to its high affinity for actin. Anantin is a RiPP used in cell biology as an atrial natriuretic peptide receptor inhibitor.[3]
In 2012-2013, a derivatized RiPP in clinical trials was LFF571. Phase II clinical trials of LFF571, a derivative of the thiopeptide GE2270-A, for the treatment of Clostridium difficile infections, with comparable safety and efficacy to vancomycin, was terminated early as the results were unfavorable.[4][5] Also recently in clinical trials was the NVB302 (a derivative of the lantibiotic actagardine) which is used for the treatment of Clostridium difficile infection.[6] Duramycin has completed phase II clinical trials for the treatment of cystic fibrosis.[7]
Other bioactive RiPPs include the antibiotics cyclothiazomycin and bottromycin, the ultra-narrow spectrum antibiotic plantazolicin, and the cytotoxin patellamide A. Streptolysin S, the toxic virulence factor of Streptococcus pyogenes, is also a RiPP. Additionally, human thyroid hormone itself is a RiPP due to its biosynthetic origin as thyroglobulin.
Classifications
Amatoxins and phallotoxins
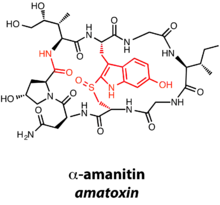
Amatoxins and phallotoxins are 8- and 7-membered natural products, respectively, characterized by N-to-C cyclization in addition to a tryptathionine motif derived from the crosslinking of Cys and Trp.[8][9] The amatoxins and phallotoxins also differ from other RiPPs based on the presence of a C-terminal recognition sequence in addition to the N-terminal leader peptide. α-Amanitin, an amatoxin, has a number of posttranslational modifications in addition to macrocyclization and formation of the tryptathionine bridge: oxidation of the tryptathionine leads to the presence of a sulfoxide, and numerous hydroxylations decorate the natural product. As an amatoxin, α-amanitin is an inhibitor of RNA polymerase II.[10]
Bottromycins
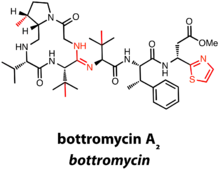
Bottromycins contain a C-terminal decarboxylated thiazole in addition to a macrocyclic amidine.[11]
There are currently six known bottromycin compounds, which differ in the extent of side chain methylation, an additional characteristic of the bottromycin class. The total synthesis of bottromycin A2 was required to definitively determine the structure of the first bottromycin.[11]
Thus far, gene clusters predicted to produce bottromycins have been identified in the genus Streptomyces. Bottromycins differ from other RiPPs in that there is no N-terminal leader peptide. Rather, the precursor peptide has a C-terminal extension of 35-37 amino acids, hypothesized to act as a recognition sequence for posttranslational machinery.[12]
Cyanobactins
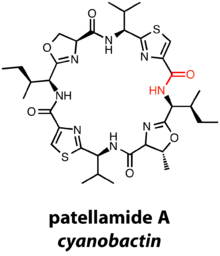
Cyanobactins are diverse metabolites from cyanobacteria with N-to-C macrocylization of a 6–20 amino acid chain. Cyanobactins are natural products isolated from cyanobacteria, and close to 30% of all cyanobacterial strains are thought to contain cyanobacterial gene clusters.[13] However, while thus far all cyanobactins are credited to cyanobacteria, there exists the possibility that other organisms could produce similar natural products.
The precursor peptide of the cyanobactin family is traditionally designated the "E" gene, whereas precursor peptides are designated gene "A" in most RiPP gene clusters. "A" is a serine protease involved in cleavage of the leader peptide and subsequent macrocyclization of the peptide natural product, in combination with an additional serine protease homologue, the encoded by gene "G". Members of the cyanobactin family may bear thiazolines/oxazolines, thiazoles/oxazoles, and methylations depending on additional modification enzymes. For example, perhaps the most famous cyanobactin is patellamide A, which contains two thiazoles, a methyloxazoline, and an oxazoline in its final state, a macrocycle derived from 8 amino acids.
Lanthipeptides
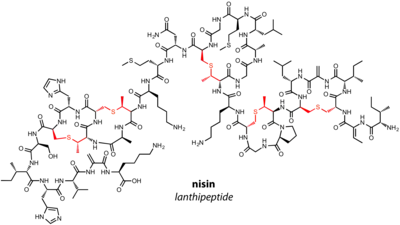
Lanthipeptides are one of the most well-studied families of RiPPs. The family is characterized by the presence of lanthionine (Lan) and 3-methyllanthionine (MeLan) residues in the final natural product. There are four major classes of lanthipeptides, delineated by the enzymes responsible for installation of Lan and MeLan. The dehydratase and cyclase can be two separate proteins or one multifunctional enzyme. Previously, lanthipeptides were known as "lantipeptides" before a consensus was reached in the field.[1]
Lantibiotics are lanthipeptides that have known antimicrobial activity. The founding member of the lanthipeptide family, nisin, is a lantibiotic that has been used to prevent the growth of food-born pathogens for over 40 years.[14]
Lasso peptides
Lasso peptides are short peptides containing an N-terminal macrolactam macrocycle "ring" through which a linear C-terminal "tail" is threaded.[15][16] Because of this threaded-loop topology, these peptides resemble lassos, giving rise to their name. They are a member of a larger class of amino-acid-based lasso structures. Additionally, lasso peptides are formally rotaxanes.
The N-terminal "ring" can be from 7 to 9 amino acids long and is formed by an isopeptide bond between the N-terminal amine of the first amino acid of the peptide and the carboxylate side chain of an aspartate or glutamate residue. The C-terminal "tail" ranges from 7 to 15 amino acids in length.[15]
The first amino acid of lasso peptides is almost invariably glycine or cysteine, with mutations at this site not being tolerated by known enzymes.[16] Thus, bioinformatics-based approaches to lasso peptide discovery have thus used this as a constraint.[15] However, some lasso peptides were recently discovered that also contain serine or alanine as their first residue.[17]
The threading of the lasso tail is trapped either by disulfide bonds between ring and tail cysteine residues (class I lasso peptides), by steric effects due to bulky residues on the tail (class II lasso peptides), or both (class III lasso peptides).[16] The compact structure makes lasso peptides frequently resistant to proteases or thermal unfolding.[16]
Linear azol(in)e-containing peptides
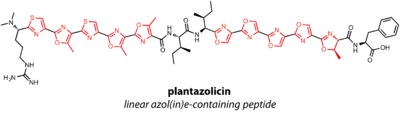
Linear azole(in)e-containing peptides (LAPs) contain thiazoles and oxazoles, or their reduced thiazoline and oxazoline forms. Thiazol(in)es are the result of cyclization of Cys residues in the precursor peptide, while (methyl)oxazol(in)es are formed from Thr and Ser. Azole and azoline formation also modifies the residue in the -1 position, or directly C-terminal to the Cys, Ser, or Thr. A dehydrogenase in the LAP gene cluster is required for oxidation of azolines to azoles.
Plantazolicin is a LAP with extensive cyclization. Two sets of five heterocycles endow the natural product with structural rigidity and unusually selective antibacterial activity.[18] Streptolysin S (SLS) is perhaps the most well-studied and most famous LAP, in part because the structure is still unknown since the discovery of SLS in 1901. Thus, while the biosynthetic gene cluster suggests SLS is a LAP, structural confirmation is lacking.
Microcins
Microcins are all RiPPs produced by Enterobacteriaceae with a molecular weight <10 kDa. Many members of other RiPP families, such as microcin E492,[19] microcin B17 (LAP) and microcin J25 (Lasso peptide) are also considered microcins. Instead of being classified based on posttranslational modifications or modifying enzymes, microcins are instead identified by molecular weight, native producer, and antibacterial activity. Microcins are either plasmid- or chromosome-encoded, but specifically have activity against Enerobacteriaceae. Because these organisms are also often producers of microcins, the gene cluster contains not only a precursor peptide and modification enzymes, but also a self-immunity gene to protect the producing strain, and genes encoding export of the natural product.
Microcins have bioactivity against Gram-negative bacteria but usually display narrow-spectrum activity due to hijacking of specific receptors involved in the transport of essential nutrients.
Thiopeptides

Most of the characterized thiopeptides have been isolated from Actinobacteria.[20] General structural features of thiopeptide macrocycles, are dehydrated amino acids and thiazole rings formed from dehydrated serine/threonine and cyclized cysteine residues, respectively
The thiopeptide macrocycle is closed with a six-membered nitrogen-bearing ring. Oxidation state and substitution pattern of the nitrogenous ring determines the series of the thiopeptide natural product.[1] While the mechanism of macrocyclization is not known, the nitrogenous ring can exist in thiopeptides as a piperidine, dehydropiperidine, or a fully oxidized pyridine. Additionally, some thiopeptides bear a second macrocycle, which bears a quinaldic acid or indolic acid residue derived from tryptophan. Perhaps the most well-characterized thiopeptide, thiostrepton A, contains a dehydropiperidine ring and a second, quinaldic acid-containing macrocycle. Four residues are dehydrated during posttranslational modification, and the final natural product also bears four thiazoles and one azoline.
Other RiPPs
Autoinducing Peptides (AIPs) and quorum sensing peptides are used as signaling molecules in the process called quorum sensing. AIPs are characterized by the presence of a cyclic ester or thioester, unlike other regulatory peptides that are linear. In pathogens, exported AIPs bind to extracellular receptors that trigger the production of virulence factors.[21] In Staphylococcus aureus, AIPs are biosynthesized from a precursor peptide composed of a C-terminal leader region, the core region, and negatively charged tail region that is, along with the leader peptide, cleaved before AIP export.[22]
Bacterial Head-to-Tail Cyclized Peptides refers exclusively to ribosomally synthesized peptides with 35-70 residues and a peptide bond between the N- and C-termini, sometimes referred to as bacteriocins, although this term is used more broadly. The distinctive nature of this class is not only the relatively large size of the natural products but also the modifying enzymes responsible for macrocyclization. Other N-to-C cyclized RiPPs, such as the cyanobactins and orbitides, have specialized biosynthetic machinery for macrocylization of much smaller core peptides. Thus far, these bacteriocins have been identified only in Gram-positive bacteria. Enterocin AS-48 was isolated from Enterococcus and, like other bacteriocins, is relatively resistant to high temperature, pH changes, and many proteases as a result of macrocyclization.[23] Based on solution structures and sequence alignments, bacteriocins appear to take on similar 3D structures despite little sequence homology, contributing to stability and resistance to degradation.
Conopeptides and other toxoglossan peptides are the components of the venom of predatory marine snails, such as the cone snails or Conus.[24] Venom peptides from cone snails are generally smaller than those found in other animal venoms (10-30 amino acids vs. 30-90 amino acids) and have more disulfide crosslinks.[24] A single species may have 50-200 conopeptides encoded in its genome, recognizable by a well-conserved signal sequence.[1]
Cyclotides are RiPPs with a head-to-tail cyclization and three conserved disulfide bonds that form a knotted structure called a cyclic cysteine knot motif.[25][26] No other posttranslational modifications have been observed on the characterized cyclotides, which are between 28 - 37 amino acids in size. Cyclotides are plant natural products and the different cyclotides appear to be species-specific. While many activities have been reported for cyclotides, it has been hypothesized that all are united by a common mechanism of binding to and disrupting the cell membrane.[27]
Glycocins are RiPPS that are glycosylated antimicrobial peptides. Only two members have been fully characterized, making this a small RiPP class.[28][29] Sublancin 168 and glycocin F are both Cys-glycosylated and, in addition, have disulfide bonds between non-glycosylated Cys residues. While both members bear S-glycosyl groups, RiPPs bearing O- or N-linked carbohydrates will also be included in this family as they are discovered.
Linaridins are characterized by C-terminal aminovinyl cysteine residues. While this posttranslational modification is also seen in the lanthipeptides epidermin and mersacidin, linaridins do not have Lan or MeLan residues. In addition, the linaridin moiety is formed from modification of two Cys residues, whereas lanthipeptide aminovinyl cysteines are formed from Cys and dehydroalanine (Dha).[30] The first linaridin to be characterized was cypemycin.[31]
Microviridins are cyclic N-acetylated trideca- and tetradecapeptides with ω-ester and/or ω-amide bonds. Lactone formation through glutamate or aspartate ω-carboxy groups and the lysine ε-amino group forms macrocycles in the final natural product.
Orbitides are plant-derived N-to-C cyclized peptides with no disulfide bonds. Also referred to as Caryophyllaceae-like homomonocyclopeptides,[32] orbitides are 5-12 amino acids in length and are composed of mainly hydrophobic residues. Similar to the amatoxins and phallotoxins, the gene sequences of orbitides suggest the presence of a C-terminal recognition sequence. In the flaxseed variety Linum usitatissimum, a precursor peptide was found using Blast searching that potentially contains five core peptides separated by putative recognition sequences.[33]
Proteusins are named after "Proteus", a Greek shape-shifting sea god. Until now, the only known members in the family of Proteusins are called polytheonamides. They were originally presumed to be nonribosomal natural products due to the presence of many D-amino acids and other non-proteinogenic amino acids. However, a metagenomic study revealed the natural products as the most extensively modified class of RiPPs known to date.[34] Six enzymes are responsible for installing a total of 48 posttranslational modifications onto the polytheonamide A and B precursor peptides, including 18 epimerizations. Polytheonamides are exceptionally large, as a single molecule is able to span a cell membrane and form an ion channel.[35][36]
Sactipeptides contain intramolecular linkages between the sulfur of Cys residues and the α-carbon of another residue in the peptide. A number of nonribosomal peptides bear the same modification. In 2003, the first RiPP with a sulfur-to-α-carbon linkage was reported when the structure of subtilosin A was determined using isotopically enriched media and NMR spectroscopy.[37] In the case of subtilosin A, isolated from Bacillus subtilis 168, the Cα crosslinks between Cys4 and Phe31, Cys7 and Thr28, and Cys13 and Phe22 are not the only posttranslational modifications; the C- and N-termini form an amide bond, resulting in a circular structure that is conformationally restricted by the Cα bonds. Sactipeptides with antimicrobial activity are commonly referred to as sactibiotics (sulfur to alpha-carbon antibiotic).[38]
Biosynthesis
RiPPs are characterized by a common biosynthetic strategy wherein genetically-encoded peptides undergo translation and subsequent chemical modification by biosynthetic enzymes.
Common features
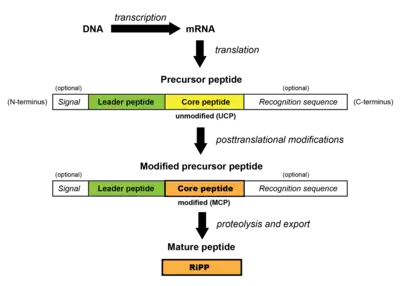
All RiPPs are synthesized first at the ribosome as a precursor peptide. This peptide consists of a core peptide segment which is typically preceded (and occasionally followed) by a leader peptide segment and is typically ~20-110 residues long. The leader peptide is usually important for enabling enzymatic processing of the precursor peptide via aiding in recognition of the core peptide by biosynthetic enzymes and for cellular export. Some RiPPs also contain a recognition sequence C-terminal to the core peptide; these are involved in excision and cyclization. Additionally, eukaryotic RiPPs may contain a signal segment of the precursor peptide which helps direct the peptide to cellular compartments.[1]
During RiPP biosynthesis, the unmodified precursor peptide (containing an unmodified core peptide, UCP) is recognized and chemically modified sequentially by biosynthetic enzymes (PRPS). Examples of modifications include dehydration (i.e. lanthipeptides, thiopeptides), cyclodehydration (i.e. thiopeptides), prenylation (i.e. cyanobactins), and cyclization (i.e. lasso peptides), among others. The resulting modified precursor peptide (containing a modified core peptide, MCP) then undergoes proteolysis, wherein the non-core regions of the precursor peptide are removed. This results in the mature RiPP.[1]
Nomenclature
Papers published prior to a recent community consensus[1] employ differing sets of nomenclature. The precursor peptide has been referred to previously as prepeptide, prepropeptide, or structural peptide. The leader peptide has been referred to as a propeptide, pro-region, or intervening region. Historical alternate terms for core peptide included propeptide, structural peptide, and toxin region (for conopeptides, specifically).[1]
Family-specific features
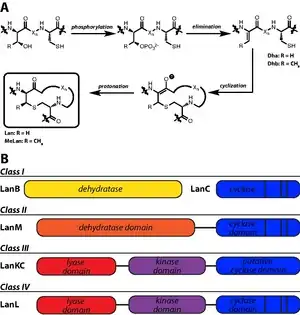
Lanthipeptides
Lanthipeptides are characterized by the presence lanthionine (Lan) and 3-methyllanthionine (MeLan) residues. Lan residues are formed from a thioether bridge between Cys and Ser, while MeLan residues are formed from the linkage of Cys to a Thr residue. The biosynthetic enzymes responsible for Lan and MeLan installation first dehydrate Ser and Thr to dehydroalanine (Dha) and dehydrobutyrine (Dhb), respectively. Subsequent thioether crosslinking occurs through a Michael-type addition by Cys onto Dha or Dhb.[39]
Four classes of lanthipeptide biosynthetic enzymes have been designated.[40] Class I lanthipeptides have dedicated lanthipeptide dehydratases, called LanB enzymes, though more specific designations are used for particular lanthipeptides (e.g. NisB is the nisin dehydratase). A separate cyclase, LanC, is responsible for the second step in Lan and MeLan biosynthesis. However, class II, III, and IV lanthipeptides have bifunctional lanthionine synthetases in their gene clusters, meaning a single enzyme carries out both dehydration and cyclization steps. Class II synthetases, designated LanM synthetases, have N-terminal dehydration domains with no sequence homology to other lanthipeptide biosynthetic enzymes; the cyclase domain has homology to LanC. Class III (LanKC) and IV (LanL) enzymes have similar N-terminal lyase and central kinase domains, but diverge in C-terminal cyclization domains: the LanL cyclase domain is homologous to LanC, but the class III enzymes lack Zn-ligand binding domains.[41]
Linear azol(in)e-containing peptides
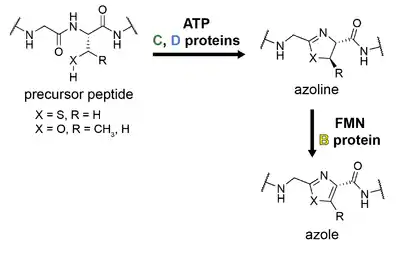
The hallmark of linear azol(in)e-containing peptide (LAP) biosynthesis is the formation of azol(in)e heterocycles from the nucleophilic amino acids serine, threonine, or cysteine.[1][42] This is accomplished by three enzymes referred to as the B, C, and D proteins; the precursor peptide is referred to as the A protein, as in other classes.[1]
The C protein is mainly involved in leader peptide recognition and binding and is sometimes called a scaffolding protein. The D protein is an ATP-dependent cyclodehydratase that catalyzes the cyclodehydration reaction, resulting in formation of an azoline ring. This occurs by direct activation of the amide backbone carbonyl with ATP, resulting in stoichiometric ATP consumption.[43] The C and D proteins are occasionally present as a single, fused protein, as is the case for trunkamide biosynthesis. The B protein is a flavin mononucleotide (FMN)-dependent dehydrogenase which oxidizes certain azoline rings into azoles.
The B protein is typically referred to as the dehydrogenase; the C and D proteins together form the cyclodehydratase, although the D protein alone performs the cyclodehydration reaction. Early work on microcin B17 adopted a different nomenclature for these proteins, but a recent consensus has been adopted by the field as described above.[1]
Cyanobactins
Cyanobactin biosynthesis requires proteolytic cleavage of both N-terminal and C-terminal portions of the precursor peptide. The defining proteins are thus an N-terminal protease, referred to as the A protein, and a C-terminal protease, referred to as the G protein. The G protein is also responsible for macrocyclization.
For cyanobactins, the precursor peptide is referred to as the E peptide.[1] Minimally, the E peptide requires a leader peptide region, a core (structural) region, and both N-terminal and C-terminal protease recognition sequences. In contrast to most RiPPs, for which a single precursor peptide encodes a single natural product via a lone core peptide, cyanobactin E peptides can contain multiple core regions; multiple E peptides can even be present in a single gene cluster.[1][44]
Many cyanobactins also undergo heterocyclization by a heterocyclase (referred to as the D protein), installing oxazoline or thiazoline moieties from Ser/Thr/Cys residues prior to the action of the A and G proteases.[1] The heterocyclase is an ATP-dependent YcaO homologue that behaves biochemically in the same manner as YcaO-domain cyclodehydratases in thiopeptide and linear azol(in)e-containing peptide (LAP) biosynthesis (described above).
A common modification is prenylation of hydroxyl groups by an F protein prenyltransferase. Oxidation of azoline heterocycles to azoles can also be accomplished by an oxidase domain located on the G protein. Unusual for ribosomal peptides, cyanobactins can include D-amino acids; these can occur adjacent to azole or azoline residues.[1] The functions of some proteins found commonly in cyanobactin biosynthetic gene clusters, the B and C proteins, are unknown.
Thiopeptides
Thiopeptide biosynthesis involves particularly extensive modification of the core peptide scaffold. Indeed, due to the highly complex structures of thiopeptides, it was commonly thought that these natural products were nonribosomal peptides. Recognition of the ribosomal origin of these molecules came in 2009 with the independent discovery of the gene clusters for several thiopeptides.[1][45][46][47][48]
The standard nomenclature for thiopeptide biosynthetic proteins follows that of the thiomuracin gene cluster.[1][47] In addition to the precursor peptide, referred to as the A peptide, thiopeptide biosynthesis requires at least six genes. These include lanthipeptide-like dehydratases, designated the B and C proteins, which install dehydroalanine and dehydrobutyrine moieties by dehydrating Ser/Thr precursor residues. Azole and azoline synthesis is effected by the E protein, the dehydrogenase, and the G protein, the cyclodehydratase. The nitrogen-containing heterocycle is installed by the D protein cyclase via a putative [4+2] cycloaddition of dehydroalanine moieties to form the characteristic macrocycle.[49] The F protein is responsible for binding of the leader peptide.[50]
Thiopeptide biosynthesis is biochemically similar to that of cyanobactins, lanthipeptides, and linear azol(in)e-containing peptides (LAPs). As with cyanobactins and LAPs, azole and azoline synthesis occurs via the action of an ATP-dependent YcaO-domain cyclodehydratase. In contrast to LAPs, where cyclodehydration occurs via the action of two distinct proteins responsible for leader peptide binding and cyclodehydrative catalysis, these are fused into a single protein (G protein) in cyanobactin and thiopeptide biosynthesis.[1] However, in thiopeptides, an additional protein, designated the Ocin-ThiF-like protein (F protein) is necessary for leader peptide recognition and potentially recruiting other biosynthetic enzymes.[50]
Lasso peptides
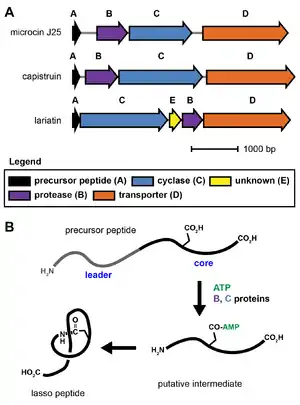
Lasso peptide biosynthesis requires at least three genes, referred to as the A, B, and C proteins.[1][15] The A gene encodes the precursor peptide, which is modified by the B and C proteins into the mature natural product. The B protein is an adenosine triphosphate-dependent cysteine protease that cleaves the leader region from the precursor peptide. The C protein displays homology to asparagine synthetase and is thought to activate the carboxylic acid side chain of a glutamate or aspartate residue via adenylylation. The N-terminal amine formed by the B protein (protease) then reacts with this activated side chain to form the macrocycle-forming isopeptide bond. The exact steps and reaction intermediates in lasso peptide biosynthesis remain unknown due to experimental difficulties associated with the proteins.[15] Commonly, the B protein is referred to as the lasso protease, and the C protein is referred to as the lasso cyclase.
Some lasso peptide biosynthetic gene clusters also require an additional protein of unknown function for biosynthesis. Additionally, lasso peptide gene clusters usually include an ABC transporter (D protein) or an isopeptidase, although these are not strictly required for lasso peptide biosynthesis and are sometimes absent.[15] No X-ray crystal structure is yet known for any lasso peptide biosynthetic protein.
The biosynthesis of lasso peptides is particularly interesting due to the inaccessibility of the threaded-lasso topology to chemical peptide synthesis.
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA (January 2013). "Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature". Natural Product Reports. 30 (1): 108–60. doi:10.1039/c2np20085f. PMC 3954855. PMID 23165928.
- ↑ Velásquez JE, van der Donk WA (2011). "Genome mining for ribosomally synthesized natural products". Current Opinion in Chemical Biology. 15 (1): 11–21. doi:10.1016/j.cbpa.2010.10.027. PMC 3090663. PMID 21095156.
- ↑ Wyss DF, Lahm HW, Manneberg M, Labhardt AM (1991). "Anantin--a peptide antagonist of the atrial natriuretic factor (ANF). II. Determination of the primary sequence by NMR on the basis of proton assignments". The Journal of Antibiotics. 44 (2): 172–80. doi:10.7164/antibiotics.44.172. PMID 1826288.
- ↑ Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, Praestgaard J, Leeds JA, Blais J, Pertel P (2015). "Multicenter, randomized clinical trial to compare the safety and efficacy of LFF571 and vancomycin for Clostridium difficile infections". Antimicrobial Agents and Chemotherapy. 59 (3): 1435–40. doi:10.1128/AAC.04251-14. PMC 4325808. PMID 25534727.
- ↑ "Safety and Efficacy of Multiple Daily Dosing of Oral LFF571 in Patients With Moderate Clostridium Difficile Infections". Retrieved 2015-06-08.
- ↑ "Assessment of the safety and distribution of NVB302 in healthy volunteers". ISRCTNregistry. 2012-10-23. Retrieved 2015-06-08.
- ↑ Sandiford SK (2015). "Perspectives on lantibiotic discovery - where have we failed and what improvements are required?". Expert Opinion on Drug Discovery. 10 (4): 315–20. doi:10.1517/17460441.2015.1016496. PMID 25697059.
- ↑ Zanotti G, Beijer B, Wieland T (September 1987). "Synthesis of cyclic tryptathionine peptides". Int. J. Pept. Protein Res. 30 (3): 323–9. doi:10.1111/j.1399-3011.1987.tb03338.x. PMID 3692680.
- ↑ Wieland T, Faulstich H (December 1978). "Amatoxins, phallotoxins, phallolysin, and antamanide: the biologically active components of poisonous Amanita mushrooms". CRC Crit. Rev. Biochem. 5 (3): 185–260. doi:10.3109/10409237809149870. PMID 363352.
- ↑ Bushnell DA, Cramer P, Kornberg RD (February 2002). "Structural basis of transcription: alpha-amanitin-RNA polymerase II cocrystal at 2.8 A resolution". Proc. Natl. Acad. Sci. U.S.A. 99 (3): 1218–22. Bibcode:2002PNAS...99.1218B. doi:10.1073/pnas.251664698. PMC 122170. PMID 11805306.
- 1 2 Shimamura H, Gouda H, Nagai K, et al. (2009). "Structure determination and total synthesis of bottromycin A2: a potent antibiotic against MRSA and VRE". Angew. Chem. Int. Ed. Engl. 48 (5): 914–7. doi:10.1002/anie.200804138. PMID 19115340.
- ↑ Gomez-Escribano JP, Song L, Bibb MJ, Challis GL (2012). "Posttranslational β-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics". Chem. Sci. 3 (12): 3522–5. doi:10.1039/C2SC21183A.
- ↑ Leikoski N, Fewer DP, Sivonen K (February 2009). "Widespread occurrence and lateral transfer of the cyanobactin biosynthesis gene cluster in cyanobacteria". Appl. Environ. Microbiol. 75 (3): 853–7. Bibcode:2009ApEnM..75..853L. doi:10.1128/AEM.02134-08. PMC 2632131. PMID 19047393.
- ↑ Lubelski J, Rink R, Khusainov R, Moll GN, Kuipers OP (2008). "Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin". Cellular and Molecular Life Sciences. 65 (3): 455–76. doi:10.1007/s00018-007-7171-2. PMID 17965835. S2CID 9549591.
- 1 2 3 4 5 6 Maksimov MO, Link AJ (February 2014). "Prospecting genomes for lasso peptides". Journal of Industrial Microbiology & Biotechnology. 41 (2): 333–44. doi:10.1007/s10295-013-1357-4. PMID 24142336. S2CID 13286422.
- 1 2 3 4 Maksimov MO, Pan SJ, James Link A (September 2012). "Lasso peptides: structure, function, biosynthesis, and engineering". Natural Product Reports. 29 (9): 996–1006. doi:10.1039/c2np20070h. PMID 22833149.
- ↑ Zimmermann, M.; Hegemann, J. D.; Xie, X.; Marahiel, M. A. (2014). "Characterization of caulonodin lasso peptides revealed unprecedented N-terminal residues and a precursor motif essential for peptide maturation". Chem. Sci. 5 (10): 4032–4043. doi:10.1039/C4SC01428F.
- ↑ Molohon KJ, Melby JO, Lee J, Evans BS, Dunbar KL, Bumpus SB, Kelleher NL, Mitchell DA (2011). "Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics". ACS Chemical Biology. 6 (12): 1307–13. doi:10.1021/cb200339d. PMC 3241860. PMID 21950656.
- ↑ Huang, Kai; Zeng, Jianwei; Liu, Xueli; Jiang, Tianyu; Wang, Jiawei (2021-04-06). "Structure of the mannose phosphotransferase system (man-PTS) complexed with microcin E492, a pore-forming bacteriocin". Cell Discovery. 7 (1): 20. doi:10.1038/s41421-021-00253-6. ISSN 2056-5968. PMC 8021565. PMID 33820910.
- ↑ Bagly MC, Dale JW, Merritt EA, Xiong X (2005). "Thiopeptide antibiotics". Chem. Rev. 105 (2): 685–714. doi:10.1021/cr0300441. PMID 15700961.
- ↑ Thoendel M, Kavanaugh JS, Flack CE, Horswill AR (January 2011). "Peptide signaling in the staphylococci". Chem. Rev. 111 (1): 117–51. doi:10.1021/cr100370n. PMC 3086461. PMID 21174435.
- ↑ Thoendel M, Horswill AR (August 2009). "Identification of Staphylococcus aureus AgrD residues required for autoinducing peptide biosynthesis". J. Biol. Chem. 284 (33): 21828–38. doi:10.1074/jbc.M109.031757. PMC 2756194. PMID 19520867.
- ↑ Sánchez-Hidalgo M, Montalbán-López M, Cebrián R, Valdivia E, Martínez-Bueno M, Maqueda M (2011). "AS-48 bacteriocin: close to perfection". Cell. Mol. Life Sci. 68 (17): 2845–57. doi:10.1007/s00018-011-0724-4. PMID 21590312. S2CID 24837331.
- 1 2 Buczek O, Bulaj G, Olivera BM (2005). "Conotoxins and the posttranslational modification of secreted gene products". Cell. Mol. Life Sci. 62 (24): 3067–79. doi:10.1007/s00018-005-5283-0. PMID 16314929. S2CID 25647743.
- ↑ Craik DJ, Daly NL, Bond T, Waine C (1999). "Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif". J. Mol. Biol. 294 (5): 1327–36. doi:10.1006/jmbi.1999.3383. PMID 10600388.
- ↑ Saether O, Craik DJ, Campbell ID, Sletten K, Juul J, Norman DG (1995). "Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1". Biochemistry. 34 (13): 4147–58. doi:10.1021/bi00013a002. PMID 7703226.
- ↑ Huang YH, Colgrave ML, Daly NL, Keleshian A, Martinac B, Craik DJ (July 2009). "The biological activity of the prototypic cyclotide kalata b1 is modulated by the formation of multimeric pores". J. Biol. Chem. 284 (31): 20699–707. doi:10.1074/jbc.M109.003384. PMC 2742835. PMID 19491108.
- ↑ Oman TJ, Boettcher JM, Wang H, Okalibe XN, van der Donk WA (2011). "Sublancin is not a lantibiotic but an S-linked glycopeptide". Nat. Chem. Biol. 7 (2): 78–80. doi:10.1038/nchembio.509. PMC 3060661. PMID 21196935.
- ↑ Garcia De Gonzalo CV, Zhu L, Oman TJ, van der Donk WA (2014). "NMR structure of the S-linked glycopeptide sublancin 168". ACS Chem. Biol. 9 (3): 796–801. doi:10.1021/cb4008106. PMC 3985867. PMID 24405370.
- ↑ Claesen J, Bibb M (2010). "Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides". Proc. Natl. Acad. Sci. U.S.A. 107 (37): 16297–302. Bibcode:2010PNAS..10716297C. doi:10.1073/pnas.1008608107. PMC 2941285. PMID 20805503.
- ↑ Komiyama K, Otoguro K, Segawa T, Shiomi K, Yang H, Takahashi Y, Hayashi M, Otani T, Omura S (1993). "A new antibiotic, cypemycin. Taxonomy, fermentation, isolation and biological characteristics". J. Antibiot. 46 (11): 1666–71. doi:10.7164/antibiotics.46.1666. PMID 7802859.
- ↑ Tan NH, Zhou J (2006). "Plant cyclopeptides". Chem. Rev. 106 (3): 840–95. doi:10.1021/cr040699h. PMID 16522011.
- ↑ Venglat P, Xiang D, Qiu S, Stone SL, Tibiche C, Cram D, Alting-Mees M, Nowak J, Cloutier S, Deyholos M, Bekkaoui F, Sharpe A, Wang E, Rowland G, Selvaraj G, Datla R (2011). "Gene expression analysis of flax seed development". BMC Plant Biol. 11: 74. doi:10.1186/1471-2229-11-74. PMC 3107784. PMID 21529361.
- ↑ Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, Sahl HG, Matsunaga S, Piel J (2012). "Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides". Science. 338 (6105): 387–90. Bibcode:2012Sci...338..387F. doi:10.1126/science.1226121. PMID 22983711. S2CID 23925994.
- ↑ Hamada T, Matsunaga S, Fujiwara M, Fujita K, Hirota H, Schmucki R, Güntert P, Fusetani N (2010). "Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge". J. Am. Chem. Soc. 132 (37): 12941–5. doi:10.1021/ja104616z. PMID 20795624.
- ↑ Iwamoto M, Shimizu H, Muramatsu I, Oiki S (2010). "A cytotoxic peptide from a marine sponge exhibits ion channel activity through vectorial-insertion into the membrane". FEBS Lett. 584 (18): 3995–9. doi:10.1016/j.febslet.2010.08.007. PMID 20699099. S2CID 30215533.
- ↑ Kawulka KE, Sprules T, Diaper CM, Whittal RM, McKay RT, Mercier P, Zuber P, Vederas JC (2004). "Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to alpha-carbon cross-links: formation and reduction of alpha-thio-alpha-amino acid derivatives". Biochemistry. 43 (12): 3385–95. doi:10.1021/bi0359527. PMID 15035610.
- ↑ Kawulka K, Sprules T, McKay RT, Mercier P, Diaper CM, Zuber P, Vederas JC (2003). "Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to alpha-carbons of phenylalanine and threonine". J. Am. Chem. Soc. 125 (16): 4726–7. doi:10.1021/ja029654t. PMID 12696888.
- ↑ Knerr PJ, van der Donk WA (2012). "Discovery, biosynthesis, and engineering of lantipeptides". Annu. Rev. Biochem. 81: 479–505. doi:10.1146/annurev-biochem-060110-113521. PMID 22404629.
- ↑ Siezen RJ, Kuipers OP, de Vos WM (1996). "Comparison of lantibiotic gene clusters and encoded proteins" (PDF). Antonie van Leeuwenhoek. 69 (2): 171–84. doi:10.1007/bf00399422. PMID 8775977. S2CID 8887022.
- ↑ Goto Y, Li B, Claesen J, Shi Y, Bibb MJ, van der Donk WA (2010). "Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary insights". PLOS Biol. 8 (3): e1000339. doi:10.1371/journal.pbio.1000339. PMC 2843593. PMID 20351769.
- ↑ Melby JO, Nard NJ, Mitchell DA (June 2011). "Thiazole/oxazole-modified microcins: complex natural products from ribosomal templates". Current Opinion in Chemical Biology. 15 (3): 369–78. doi:10.1016/j.cbpa.2011.02.027. PMC 3947797. PMID 21429787.
- ↑ Dunbar KL, Melby JO, Mitchell DA (June 2012). "YcaO domains use ATP to activate amide backbones during peptide cyclodehydrations". Nature Chemical Biology. 8 (6): 569–75. doi:10.1038/nchembio.944. PMC 3428213. PMID 22522320.
- ↑ Donia MS, Schmidt EW (2011). "Linking chemistry and genetics in the growing cyanobactin natural products family". Chemistry & Biology. 18 (4): 508–19. doi:10.1016/j.chembiol.2011.01.019. PMC 3119926. PMID 21513887.
- ↑ Kelly WL, Pan L, Li C (2009). "Thiostrepton biosynthesis: prototype for a new family of bacteriocins". Journal of the American Chemical Society. 131 (12): 4327–34. doi:10.1021/ja807890a. PMID 19265401.
- ↑ Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA (2009). "Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin". Proceedings of the National Academy of Sciences of the United States of America. 106 (8): 2549–53. Bibcode:2009PNAS..106.2549W. doi:10.1073/pnas.0900008106. PMC 2650375. PMID 19196969.
- 1 2 Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, Hein AE, Schmitt EK, Krastel P (2009). "Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu". Journal of the American Chemical Society. 131 (16): 5946–55. doi:10.1021/ja900488a. PMID 19338336.
- ↑ Liao R, Duan L, Lei C, Pan H, Ding Y, Zhang Q, Chen D, Shen B, Yu Y, Liu W (2009). "Thiopeptide biosynthesis featuring ribosomally synthesized precursor peptides and conserved posttranslational modifications". Chemistry & Biology. 16 (2): 141–7. doi:10.1016/j.chembiol.2009.01.007. PMC 2676563. PMID 19246004.
- ↑ Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, Bowers AA (2015). "Chemoenzymatic synthesis of thiazolyl peptide natural products featuring an enzyme-catalyzed formal [4 + 2] cycloaddition". Journal of the American Chemical Society. 137 (10): 3494–7. doi:10.1021/jacs.5b00940. PMC 4425689. PMID 25742119.
- 1 2 Dunbar KL, Tietz JI, Cox CL, Burkhart BJ, Mitchell DA (2015). "Identification of an Auxiliary Leader Peptide-Binding Protein Required for Azoline Formation in Ribosomal Natural Products". Journal of the American Chemical Society. 137 (24): 7672–7. doi:10.1021/jacs.5b04682. PMC 4481143. PMID 26024319.