In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes (observable characteristics) to genotypes (the genetic constitution of organisms), uncovering genetic associations.[1][2]
Theory
Association mapping is based on the idea that traits that have entered a population only recently will still be linked to the surrounding genetic sequence of the original evolutionary ancestor, or in other words, will more often be found within a given haplotype, than outside of it. It is most often performed by scanning the entire genome for significant associations between a panel of single nucleotide polymorphisms (SNPs) (which, in many cases are spotted onto glass slides to create "SNP chips") and a particular phenotype. These associations must then be independently verified in order to show that they either (a) contribute to the trait of interest directly, or (b) are linked to/ in linkage disequilibrium with a quantitative trait locus (QTL) that contributes to the trait of interest.[3]
Association mapping seeks to identify specific functional genetic variants (loci, alleles) linked to phenotypic differences in a trait to facilitate detection of trait causing DNA sequence polymorphisms and selection of genotypes that closely resemble the phenotype. In order to identify these functional variants, it requires high throughput markers like SNPs.[4]
Use
The advantage of association mapping is that it can map quantitative traits with high resolution in a way that is statistically very powerful. Association mapping, however, also requires extensive knowledge of SNPs within the genome of the organism of interest, and is therefore difficult to perform in species that have not been well studied or do not have well-annotated genomes.[5] Association mapping has been most widely applied to the study of human disease, specifically in the form of a genome-wide association study (GWAS). A genome-wide association study is performed by scanning an entire genome for SNPs associated with a particular trait of interest, or in the case of human disease, with a particular disease of interest.[3][6] To date, thousands of genome wide associations studies have been performed on the human genome in an attempt to identify SNPs associated with a wide variety of complex human diseases (e.g. cancer, Alzheimer's disease, and obesity). The results of all such published GWAS are maintained in an NIH database (figure 1). Whether or not these studies have been clinically and/or therapeutically useful, however, remains controversial.[6]
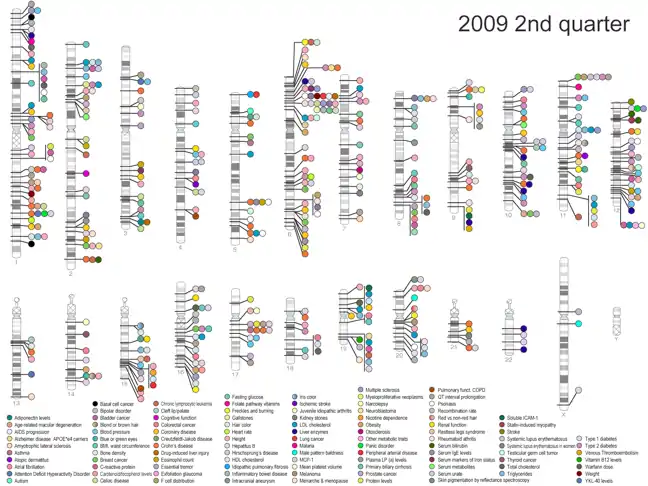
Types and variations
(A) Association mapping in population where members are assumed to be independent.
Several standard methods to test for association. Case control studies – Case control studies was among the first approaches utilized to determine whether particular genetic variant is associated with increased risk of disease in humans. Woofle, in 1955, proposed a relative risk statistic that could be used to assess genotype dependent risk. However persistent concern regarding these studies is the adequacy of matching cases and controls. In particular, population stratification can produce false positive associations. In response to this concern, Falk and Rubenstein (1987) suggested a method for assessing relative risk that uses family based controls, obviating this source of potential error. Basically, the method uses a control sample of the parental alleles or haplotypes not transmitted to affected offspring.
(B) Association mapping population where members are assumed to be related
In the real world it is very hard to find independent (unrelated) individuals. Population based association mapping has been modified to control population stratification or relatedness in nested association mapping. Still there is one other limitation in population based QTL mapping; when the frequency of the favorable allele should be relatively high to be detected. Usually favorable alleles are rare mutant alleles (for example usually a resistant parent might be 1 out of 10000 genotypes). Another variant of association mapping in related populations is family based association mapping. In family based association mapping instead of multiple unrelated individuals multiple unrelated families or pedigrees are used. The family-based association mapping[7] can be used in situations where the mutant alleles have been introgressed in populations. One popular family-based association mapping is the transmission disequilibrium test. For details, see Family based QTL mapping.
Advantages
The advantages of population based association mapping, utilizing a sample of individuals from the germplasm collections or a natural population, over traditional QTL-mapping in biparental crosses, primarily are due to availability of broader genetic variations with wider background for marker and trait correlations. The advantage of association mapping is that it can map quantitative traits with high resolution in a way that is statistically very powerful. The resolution of the mapping depends on the extent of LD, or non-random association of markers, that has occurred across the genome. Association mapping offers the opportunity to investigate diverse genetic material and potentially identify multiple alleles and mechanisms of underlying traits. It uses recombination events that have occurred over an extended period of time. Association mapping allows the possibility of exploiting historically measured trait data for association, and lastly has no need for the development of expensive and tedious biparental populations that makes approach timesaving and cost-effective.[8][9]
Limitations
A major issue with association studies is a tendency to find false positives. Populations showing a desired trait also carry a specific gene variant not because the variant actually controls the trait, but due to genetic relatedness. In particular, indirect associations that are not causal will not be eliminated by increasing the sample size or the number of markers. The main sources of such false positives are linkage between causal and noncausal sites, more than one causal site and epistasis. These indirect associations are not randomly distributed throughout the genome and are less common than false positives arising from population structure.[10]
Likewise, population structure has always remained a consistent issue. Population structure leads to spurious associations between markers and the trait. This generally is not a problem in linkage analysis because researchers know the genetic structure of the family they created. But in association mapping, where relationships between diverse populations are not necessarily well understood, marker–trait associations arising from kinship and evolutionary history can easily be mistaken for causal ones. This can be accounted for with mixed models MLM. Also called the Q+K model, it was developed to further reduce the false positive rate by controlling for both population structure and cryptic familial relatedness.[11]
See also
References
- ↑ Breseghello, Flavio; Sorrells, Mark E (2006-02-01). "Association Mapping of Kernel Size and Milling Quality in Wheat ( Triticum aestivum L.) Cultivars". Genetics. 172 (2): 1165–1177. doi:10.1534/genetics.105.044586. ISSN 1943-2631. PMC 1456215. PMID 16079235.
- ↑ Zondervan, Krina T.; Cardon, Lon R. (2007-02-01). "Designing candidate gene and genome-wide case–control association studies". Nature Protocols. 2 (10): 2492–2501. doi:10.1038/nprot.2007.366. ISSN 1750-2799. PMC 4180089. PMID 17947991.
- 1 2 Gibson, G.; Muse S.V. (2009). A Primer of Genome Science. MA: Sinauer Associates.
- ↑ Hoeschele, I. (2004-07-15). "Mapping Quantitative Trait Loci in Outbred Pedigrees". Handbook of Statistical Genetics. Chichester: John Wiley & Sons, Ltd. doi:10.1002/0470022620.bbc17. ISBN 978-0470022627.
- ↑ Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. (2008). "Genetic design and statistical power of nested association mapping in maize". Genetics. 178 (1): 539–551. doi:10.1534/genetics.107.074245. PMC 2206100. PMID 18202393.
- 1 2 Nussbaum, R.L.; McInnes, R.R.; Willard, H.F. (2007). Genetics in Medicine. Philadelphia, PA: Saunders Elsevier.
- ↑ Rosyara U.R., J.L. Gonzalez-Hernandez, K.D. Glover, K.R. Gedye and J.M. Stein. 2009. Family-based mapping of quantitative trait loci in plant breeding populations with resistance to Fusarium head blight in wheat as an illustration Theoretical and Applied Genetics 118:1617-1631 external link
- ↑ Abdurakhmonov, Ibrokhim Y.; Abdukarimov, Abdusattor (2008-06-08). "Application of Association Mapping to Understanding the Genetic Diversity of Plant Germplasm Resources". International Journal of Plant Genomics. Hindawi. 2008: 574927. doi:10.1155/2008/574927. ISSN 1687-5370. PMC 2423417. PMID 18551188. S2CID 7629296.
- ↑ Kraakman, A. T. W. (2004-09-01). "Linkage Disequilibrium Mapping of Yield and Yield Stability in Modern Spring Barley Cultivars". Genetics. 168 (1): 435–446. doi:10.1534/genetics.104.026831. ISSN 0016-6731. PMC 1448125. PMID 15454555.
- ↑ Platt, A.; Vilhjalmsson, B. J.; Nordborg, M. (2010-09-02). "Conditions Under Which Genome-Wide Association Studies Will be Positively Misleading". Genetics. 186 (3): 1045–1052. doi:10.1534/genetics.110.121665. ISSN 0016-6731. PMC 2975277. PMID 20813880.
- ↑ Yu, Jianming; Pressoir, Gael; Briggs, William H; Vroh Bi, Irie; Yamasaki, Masanori; Doebley, John F; McMullen, Michael D; Gaut, Brandon S; Nielsen, Dahlia M (2005-12-25). "A unified mixed-model method for association mapping that accounts for multiple levels of relatedness". Nature Genetics. 38 (2): 203–208. doi:10.1038/ng1702. ISSN 1061-4036. PMID 16380716. S2CID 8507433.