
Neurotoxins are toxins that are destructive to nerve tissue (causing neurotoxicity).[3] Neurotoxins are an extensive class of exogenous chemical neurological insults[4] that can adversely affect function in both developing and mature nervous tissue.[5] The term can also be used to classify endogenous compounds, which, when abnormally contacted, can prove neurologically toxic.[4] Though neurotoxins are often neurologically destructive, their ability to specifically target neural components is important in the study of nervous systems.[6] Common examples of neurotoxins include lead,[7] ethanol (drinking alcohol),[8] glutamate,[9] nitric oxide,[10] botulinum toxin (e.g. Botox),[11] tetanus toxin,[12] and tetrodotoxin.[6] Some substances such as nitric oxide and glutamate are in fact essential for proper function of the body and only exert neurotoxic effects at excessive concentrations.
Neurotoxins inhibit neuron control over ion concentrations across the cell membrane,[6] or communication between neurons across a synapse.[13] Local pathology of neurotoxin exposure often includes neuron excitotoxicity or apoptosis[14] but can also include glial cell damage.[15] Macroscopic manifestations of neurotoxin exposure can include widespread central nervous system damage such as intellectual disability,[5] persistent memory impairments,[16] epilepsy, and dementia.[17] Additionally, neurotoxin-mediated peripheral nervous system damage such as neuropathy or myopathy is common. Support has been shown for a number of treatments aimed at attenuating neurotoxin-mediated injury, such as antioxidant[8] and antitoxin[18] administration.
Background
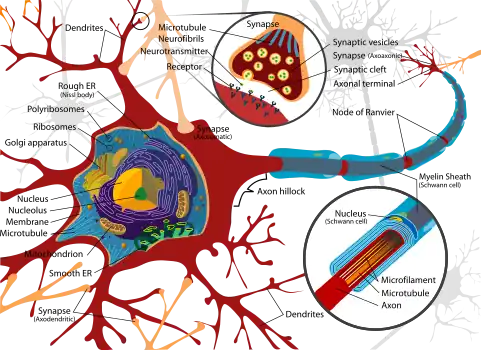
Exposure to neurotoxins in society is not new,[19] as civilizations have been exposed to neurologically destructive compounds for thousands of years. One notable example is the possible significant lead exposure during the Roman Empire resulting from the development of extensive plumbing networks and the habit of boiling vinegared wine in lead pans to sweeten it, the process generating lead acetate, known as "sugar of lead".[20] In part, neurotoxins have been part of human history because of the fragile and susceptible nature of the nervous system, making it highly prone to disruption.
The nervous tissue found in the brain, spinal cord, and periphery comprises an extraordinarily complex biological system that largely defines many of the unique traits of individuals. As with any highly complex system, however, even small perturbations to its environment can lead to significant functional disruptions. Properties leading to the susceptibility of nervous tissue include a high surface area of neurons, a high lipid content which retains lipophilic toxins, high blood flow to the brain inducing increased effective toxin exposure, and the persistence of neurons through an individual's lifetime, leading to compounding of damages.[21] As a result, the nervous system has a number of mechanisms designed to protect it from internal and external assaults, including the blood brain barrier.
The blood–brain barrier (BBB) is one critical example of protection which prevents toxins and other adverse compounds from reaching the brain.[22] As the brain requires nutrient entry and waste removal, it is perfused by blood flow. Blood can carry a number of ingested toxins, however, which would induce significant neuron death if they reach nervous tissue. Thus, protective cells termed astrocytes surround the capillaries in the brain and absorb nutrients from the blood and subsequently transport them to the neurons, effectively isolating the brain from a number of potential chemical insults.[22]
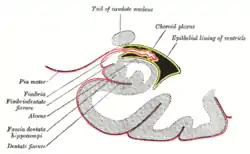
This barrier creates a tight hydrophobic layer around the capillaries in the brain, inhibiting the transport of large or hydrophilic compounds. In addition to the BBB, the choroid plexus provides a layer of protection against toxin absorption in the brain. The choroid plexuses are vascularized layers of tissue found in the third, fourth, and lateral ventricles of the brain, which through the function of their ependymal cells, are responsible for the synthesis of cerebrospinal fluid (CSF).[23] Importantly, through selective passage of ions and nutrients and trapping heavy metals such as lead, the choroid plexuses maintain a strictly regulated environment which contains the brain and spinal cord.[22][23]
By being hydrophobic and small, or inhibiting astrocyte function, some compounds including certain neurotoxins are able to penetrate into the brain and induce significant damage. In modern times, scientists and physicians have been presented with the challenge of identifying and treating neurotoxins, which has resulted in a growing interest in both neurotoxicology research and clinical studies.[24] Though clinical neurotoxicology is largely a burgeoning field, extensive inroads have been made in the identification of many environmental neurotoxins leading to the classification of 750 to 1000 known potentially neurotoxic compounds.[21] Due to the critical importance of finding neurotoxins in common environments, specific protocols have been developed by the United States Environmental Protection Agency (EPA) for testing and determining neurotoxic effects of compounds (USEPA 1998). Additionally, in vitro systems have increased in use as they provide significant improvements over the more common in vivo systems of the past. Examples of improvements include tractable, uniform environments, and the elimination of contaminating effects of systemic metabolism.[24] In vitro systems, however, have presented problems as it has been difficult to properly replicate the complexities of the nervous system, such as the interactions between supporting astrocytes and neurons in creating the BBB.[25] To even further complicate the process of determining neurotoxins when testing in-vitro, neurotoxicity and cytotoxicity may be difficult to distinguish as exposing neurons directly to compounds may not be possible in-vivo, as it is in-vitro. Additionally, the response of cells to chemicals may not accurately convey a distinction between neurotoxins and cytotoxins, as symptoms like oxidative stress or skeletal modifications may occur in response to either.[26]
In an effort to address this complication, neurite outgrowths (either axonal or dendritic) in response to applied compounds have recently been proposed as a more accurate distinction between true neurotoxins and cytotoxins in an in-vitro testing environment. Due to the significant inaccuracies associated with this process, however, it has been slow in gaining widespread support.[27] Additionally, biochemical mechanisms have become more widely used in neurotoxin testing, such that compounds can be screened for sufficiency to induce cell mechanism interference, like the inhibition of acetylcholinesterase capacity of organophosphates (includes parathion and sarin gas).[28] Though methods of determining neurotoxicity still require significant development, the identification of deleterious compounds and toxin exposure symptoms has undergone significant improvement.
Applications in neuroscience
Though diverse in chemical properties and functions, neurotoxins share the common property that they act by some mechanism leading to either the disruption or destruction of necessary components within the nervous system. Neurotoxins, however, by their very design can be very useful in the field of neuroscience. As the nervous system in most organisms is both highly complex and necessary for survival, it has naturally become a target for attack by both predators and prey. As venomous organisms often use their neurotoxins to subdue a predator or prey very rapidly, toxins have evolved to become highly specific to their target channels such that the toxin does not readily bind other targets[29] (see Ion Channel toxins). As such, neurotoxins provide an effective means by which certain elements of the nervous system may be accurately and efficiently targeted. An early example of neurotoxin based targeting used radiolabeled tetrodotoxin to assay sodium channels and obtain precise measurements about their concentration along nerve membranes.[29] Likewise through isolation of certain channel activities, neurotoxins have provided the ability to improve the original Hodgkin-Huxley model of the neuron in which it was theorized that single generic sodium and potassium channels could account for most nervous tissue function.[29] From this basic understanding, the use of common compounds such as tetrodotoxin, tetraethylammonium, and bungarotoxins have led to a much deeper understanding of the distinct ways in which individual neurons may behave.
Mechanisms of activity
As neurotoxins are compounds which adversely affect the nervous system, a number of mechanisms through which they function are through the inhibition of neuron cellular processes. These inhibited processes can range from membrane depolarization mechanisms to inter-neuron communication. By inhibiting the ability for neurons to perform their expected intracellular functions, or pass a signal to a neighboring cell, neurotoxins can induce systemic nervous system arrest as in the case of botulinum toxin,[13] or even nervous tissue death.[30] The time required for the onset of symptoms upon neurotoxin exposure can vary between different toxins, being on the order of hours for botulinum toxin[18] and years for lead.[31]
| Neurotoxin classification | Neurotoxins |
|---|---|
| Na channel inhibitors | Tetrodotoxin[6] |
| K channel inhibitors | Tetraethylammonium[32] |
| Cl channel inhibitors | Chlorotoxin,[33] |
| Ca channel inhibitors | Conotoxin[34] |
| Inhibitors of synaptic vesicle release | Botulinum toxin,[35] |
| Blood brain barrier inhibitors | Aluminium,[37] |
| Receptor inhibitors/antagonists | Bungarotoxin,[39] |
| Receptor agonists | Anatoxin-a,[41][42] |
| Cytoskeleton interference | Ammonia,[46] |
| Ca-mediated cytotoxicity | Lead[48] |
| Protein misfolding | Tau protein |
| Multiple effects | Ethanol,[49][50] |
| Receptor-selective neurotoxins | MPP+ |
| Endogenous neurotoxin sources | Nitric oxide,[52] |
Inhibitors
Sodium channel
Tetrodotoxin

Tetrodotoxin (TTX) is a poison produced by organisms belonging to the Tetraodontiformes order, which includes the puffer fish, ocean sunfish, and porcupine fish.[55] Within the puffer fish, TTX is found in the liver, gonads, intestines, and skin.[6][56] TTX can be fatal if consumed, and has become a common form of poisoning in many countries. Common symptoms of TTX consumption include paraesthesia (often restricted to the mouth and limbs), muscle weakness, nausea, and vomiting[55] and often manifest within 30 minutes of ingestion.[57] The primary mechanism by which TTX is toxic is through the inhibition of sodium channel function, which reduces the functional capacity of neuron communication. This inhibition largely affects a susceptible subset of sodium channels known as TTX-sensitive (TTX-s), which also happens to be largely responsible for the sodium current that drives the depolarization phase of neuron action potentials.[6]

TTX-resistant (TTX-r) is another form of sodium channel which has limited sensitivity to TTX, and is largely found in small diameter axons such as those found in nociception neurons.[6] When a significant level of TTX is ingested, it will bind sodium channels on neurons and reduce their membrane permeability to sodium. This results in an increased effective threshold of required excitatory signals in order to induce an action potential in a postsynaptic neuron.[6] The effect of this increased signaling threshold is a reduced excitability of postsynaptic neurons, and subsequent loss of motor and sensory function which can result in paralysis and death. Though assisted ventilation may increase the chance of survival after TTX exposure, there is currently no antitoxin. The use of the acetylcholinesterase inhibitor Neostigmine or the muscarinic acetylcholine antagonist atropine (which will inhibit parasympathetic activity), however, can increase sympathetic nerve activity enough to improve the chance of survival after TTX exposure.[55]
Potassium channel
Tetraethylammonium
Tetraethylammonium (TEA) is a compound that, like a number of neurotoxins, was first identified through its damaging effects to the nervous system and shown to have the capacity of inhibiting the function of motor nerves and thus the contraction of the musculature in a manner similar to that of curare.[58] Additionally, through chronic TEA administration, muscular atrophy would be induced.[58] It was later determined that TEA functions in-vivo primarily through its ability to inhibit both the potassium channels responsible for the delayed rectifier seen in an action potential and some population of calcium-dependent potassium channels.[32] It is this capability to inhibit potassium flux in neurons that has made TEA one of the most important tools in neuroscience. It has been hypothesized that the ability for TEA to inhibit potassium channels is derived from its similar space-filling structure to potassium ions.[58] What makes TEA very useful for neuroscientists is its specific ability to eliminate potassium channel activity, thereby allowing the study of neuron response contributions of other ion channels such as voltage gated sodium channels.[59] In addition to its many uses in neuroscience research, TEA has been shown to perform as an effective treatment of Parkinson's disease through its ability to limit the progression of the disease.[60]
Chloride channel
Chlorotoxin
Chlorotoxin (Cltx) is the active compound found in scorpion venom, and is primarily toxic because of its ability to inhibit the conductance of chloride channels.[33] Ingestion of lethal volumes of Cltx results in paralysis through this ion channel disruption. Similar to botulinum toxin, Cltx has been shown to possess significant therapeutic value. Evidence has shown that Cltx can inhibit the ability for gliomas to infiltrate healthy nervous tissue in the brain, significantly reducing the potential invasive harm caused by tumors.[61][62]
Calcium channel
Conotoxin
Conotoxins represent a category of poisons produced by the marine cone snail, and are capable of inhibiting the activity of a number of ion channels such as calcium, sodium, or potassium channels.[63][64] In many cases, the toxins released by the different types of cone snails include a range of different types of conotoxins, which may be specific for different ion channels, thus creating a venom capable of widespread nerve function interruption.[63] One of the unique forms of conotoxins, ω-conotoxin (ω-CgTx) is highly specific for Ca channels and has shown usefulness in isolating them from a system.[65] As calcium flux is necessary for proper excitability of a cell, any significant inhibition could prevent a large amount of functionality. Significantly, ω-CgTx is capable of long term binding to and inhibition of voltage-dependent calcium channels located in the membranes of neurons but not those of muscle cells.[66]
Synaptic vesicle release
Botulinum toxin
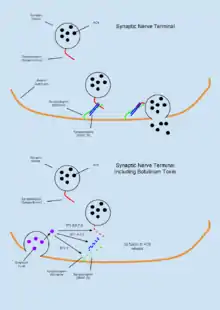
Botulinum toxin (BTX) is a group of neurotoxins consisting of eight distinct compounds, referred to as BTX-A,B,C,D,E,F,G,H, which are produced by the bacterium Clostridium botulinum and lead to muscular paralysis.[67] A notably unique feature of BTX is its relatively common therapeutic use in treating dystonia and spasticity disorders,[67] as well as in inducing muscular atrophy[11] despite being the most poisonous substance known.[18] BTX functions peripherally to inhibit acetylcholine (ACh) release at the neuromuscular junction through degradation of the SNARE proteins required for ACh vesicle-membrane fusion.[35] As the toxin is highly biologically active, an estimated dose of 1μg/kg body weight is sufficient to induce an insufficient tidal volume and resultant death by asphyxiation.[13] Due to its high toxicity, BTX antitoxins have been an active area of research. It has been shown that capsaicin (active compound responsible for heat in chili peppers) can bind the TRPV1 receptor expressed on cholinergic neurons and inhibit the toxic effects of BTX.[18]
Tetanus toxin
Tetanus neurotoxin (TeNT) is a compound that functionally reduces inhibitory transmissions in the nervous system resulting in muscular tetany. TeNT is similar to BTX, and is in fact highly similar in structure and origin; both belonging to the same category of clostridial neurotoxins.[12] Like BTX, TeNT inhibits inter-neuron communication by means of vesicular neurotransmitter (NT) release.[36] One notable difference between the two compounds is that while BTX inhibits muscular contractions, TeNT induces them. Though both toxins inhibit vesicle release at neuron synapses, the reason for this different manifestation is that BTX functions mainly in the peripheral nervous system (PNS) while TeNT is largely active in the central nervous system (CNS).[68] This is a result of TeNT migration through motor neurons to the inhibitory neurons of the spinal cord after entering through endocytosis.[69] This results in a loss of function in inhibitory neurons within the CNS resulting in systemic muscular contractions. Similar to the prognosis of a lethal dose of BTX, TeNT leads to paralysis and subsequent suffocation.[69]
Blood brain barrier
Aluminium
Neurotoxic behavior of Aluminium is known to occur upon entry into the circulatory system, where it can migrate to the brain and inhibit some of the crucial functions of the blood brain barrier (BBB).[37] A loss of function in the BBB can produce significant damage to the neurons in the CNS, as the barrier protecting the brain from other toxins found in the blood will no longer be capable of such action. Though the metal is known to be neurotoxic, effects are usually restricted to patients incapable of removing excess ions from the blood, such as those experiencing renal failure.[70] Patients experiencing aluminium toxicity can exhibit symptoms such as impaired learning and reduced motor coordination.[71] Additionally, systemic aluminium levels are known to increase with age, and have been shown to correlate with Alzheimer's disease, implicating it as a neurotoxic causative compound of the disease.[72] Despite its known toxicity in its ionic form, studies are divided on the potential toxicity of using aluminium in packaging and cooking appliances.
Mercury
Mercury is capable of inducing CNS damage by migrating into the brain by crossing the BBB.[38] Mercury exists in a number of different compounds, though methylmercury (MeHg+), dimethylmercury and diethylmercury are the only significantly neurotoxic forms. Diethylmercury and dimethylmercury are considered some of the most potent neurotoxins ever discovered.[38] MeHg+ is usually acquired through consumption of seafood, as it tends to concentrate in organisms high on the food chain.[73] It is known that the mercuric ion inhibits amino acid (AA) and glutamate (Glu) transport, potentially leading to excitotoxic effects.[74]
Receptor agonists and antagonists
Anatoxin-a
| External videos | |
|---|---|
University of Nottingham |
Investigations into anatoxin-a, also known as "Very Fast Death Factor", began in 1961 following the deaths of cows that drank from a lake containing an algal bloom in Saskatchewan, Canada.[41][42] It is a cyanotoxin produced by at least four different genera of cyanobacteria, and has been reported in North America, Europe, Africa, Asia, and New Zealand.[75]
Toxic effects from anatoxin-a progress very rapidly because it acts directly on the nerve cells (neurons). The progressive symptoms of anatoxin-a exposure are loss of coordination, twitching, convulsions and rapid death by respiratory paralysis. The nerve tissues which communicate with muscles contain a receptor called the nicotinic acetylcholine receptor. Stimulation of these receptors causes a muscular contraction. The anatoxin-a molecule is shaped so it fits this receptor, and in this way it mimics the natural neurotransmitter normally used by the receptor, acetylcholine. Once it has triggered a contraction, anatoxin-a does not allow the neurons to return to their resting state, because it is not degraded by cholinesterase which normally performs this function. As a result, the muscle cells contract permanently, the communication between the brain and the muscles is disrupted and breathing stops.[76][77]
When it was first discovered, the toxin was called the Very Fast Death Factor (VFDF) because when it was injected into the body cavity of mice it induced tremors, paralysis and death within a few minutes. In 1977, the structure of VFDF was determined as a secondary, bicyclic amine alkaloid, and it was renamed anatoxin-a.[78][79] Structurally, it is similar to cocaine.[80] There is continued interest in anatoxin-a because of the dangers it presents to recreational and drinking waters, and because it is a particularly useful molecule for investigating acetylcholine receptors in the nervous system.[81] The deadliness of the toxin means that it has a high military potential as a toxin weapon.[82]
Bungarotoxin
Bungarotoxin is a compound with known interaction with nicotinic acetylcholine receptors (nAChRs), which constitute a family of ion channels whose activity is triggered by neurotransmitter binding.[83] Bungarotoxin is produced in a number of different forms, though one of the commonly used forms is the long chain alpha form, α-bungarotoxin, which is isolated from the banded krait snake.[39] Though extremely toxic if ingested, α-bungarotoxin has shown extensive usefulness in neuroscience as it is particularly adept at isolating nAChRs due to its high affinity to the receptors.[39] As there are multiple forms of bungarotoxin, there are different forms of nAChRs to which they will bind, and α-bungarotoxin is particularly specific for α7-nAChR.[84] This α7-nAChR functions to allow calcium ion influx into cells, and thus when blocked by ingested bungarotoxin will produce damaging effects, as ACh signaling will be inhibited.[84] Likewise, the use of α-bungarotoxin can be very useful in neuroscience if it is desirable to block calcium flux in order to isolate effects of other channels. Additionally, different forms of bungarotoxin may be useful for studying inhibited nAChRs and their resultant calcium ion flow in different systems of the body. For example, α-bungarotoxin is specific for nAChRs found in the musculature and κ-bungarotoxin is specific for nAChRs found in neurons.[85]
Caramboxin
Caramboxin (CBX) is a toxin found in star fruit (Averrhoa carambola). Individuals with some types of kidney disease are susceptible to adverse neurological effects including intoxication, seizures and even death after eating star fruit or drinking juice made of this fruit. Caramboxin is a new nonpeptide amino acid toxin that stimulate the glutamate receptors in neurons. Caramboxin is an agonist of both NMDA and AMPA glutamatergic ionotropic receptors with potent excitatory, convulsant, and neurodegenerative properties.[43]
Curare
The term "curare" is ambiguous because it has been used to describe a number of poisons which at the time of naming were understood differently from present day understandings. In the past the characterization has meant poisons used by South American tribes on arrows or darts, though it has matured to specify a specific categorization of poisons which act on the neuromuscular junction to inhibit signaling and thus induce muscle relaxation.[86] The neurotoxin category contains a number of distinct poisons, though all were originally purified from plants originating in South America.[86] The effect with which injected curare poison is usually associated is muscle paralysis and resultant death.[87] Curare notably functions to inhibit nicotinic acetylcholine receptors at the neuromuscular junction. Normally, these receptor channels allow sodium ions into muscle cells to initiate an action potential that leads to muscle contraction. By blocking the receptors, the neurotoxin is capable of significantly reducing neuromuscular junction signaling, an effect which has resulted in its use by anesthesiologists to produce muscular relaxation.[88]
Cytoskeleton interference
Ammonia
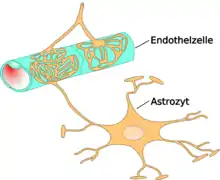
Ammonia toxicity is often seen through two routes of administration, either through consumption or through endogenous ailments such as liver failure.[89][90] One notable case in which ammonia toxicity is common is in response to cirrhosis of the liver which results in hepatic encephalopathy, and can result in cerebral edema (Haussinger 2006). This cerebral edema can be the result of nervous cell remodeling. As a consequence of increased concentrations, ammonia activity in-vivo has been shown to induce swelling of astrocytes in the brain through increased production of cGMP (Cyclic Guanosine Monophosphate) within the cells which leads to Protein Kinase G-mediated (PKG) cytoskeletal modifications.[46] The resultant effect of this toxicity can be reduced brain energy metabolism and function. Importantly, the toxic effects of ammonia on astrocyte remodeling can be reduced through administration of L-carnitine.[89] This astrocyte remodeling appears to be mediated through ammonia-induced mitochondrial permeability transition. This mitochondrial transition is a direct result of glutamine activity a compound which forms from ammonia in-vivo.[91] Administration of antioxidants or glutaminase inhibitor can reduce this mitochondrial transition, and potentially also astrocyte remodeling.[91]
Arsenic
Arsenic is a neurotoxin commonly found concentrated in areas exposed to agricultural runoff, mining, and smelting sites (Martinez-Finley 2011). One of the effects of arsenic ingestion during the development of the nervous system is the inhibition of neurite growth[92] which can occur both in PNS and the CNS.[93] This neurite growth inhibition can often lead to defects in neural migration, and significant morphological changes of neurons during development,[94]) often leading to neural tube defects in neonates.[95] As a metabolite of arsenic, arsenite is formed after ingestion of arsenic and has shown significant toxicity to neurons within about 24 hours of exposure. The mechanism of this cytotoxicity functions through arsenite-induced increases in intracellular calcium ion levels within neurons, which may subsequently reduce mitochondrial transmembrane potential which activates caspases, triggering cell death.[94] Another known function of arsenite is its destructive nature towards the cytoskeleton through inhibition of neurofilament transport.[47] This is particularly destructive as neurofilaments are used in basic cell structure and support. Lithium administration has shown promise, however, in restoring some of the lost neurofilament motility.[96] Additionally, similar to other neurotoxin treatments, the administration of certain antioxidants has shown some promise in reducing neurotoxicity of ingested arsenic.[94]
Calcium-mediated cytotoxicity
Lead

Lead is a potent neurotoxin whose toxicity has been recognized for at least thousands of years.[97] Though neurotoxic effects for lead are found in both adults and young children, the developing brain is particularly susceptible to lead-induced harm, effects which can include apoptosis and excitotoxicity.[97] An underlying mechanism by which lead is able to cause harm is its ability to be transported by calcium ATPase pumps across the BBB, allowing for direct contact with the fragile cells within the central nervous system.[98] Neurotoxicity results from lead's ability to act in a similar manner to calcium ions, as concentrated lead will lead to cellular uptake of calcium which disrupts cellular homeostasis and induces apoptosis.[48] It is this intracellular calcium increase that activates protein kinase C (PKC), which manifests as learning deficits in children as a result of early lead exposure.[48] In addition to inducing apoptosis, lead inhibits interneuron signaling through the disruption of calcium-mediated neurotransmitter release.[99]
Neurotoxins with multiple effects
Ethanol

As a neurotoxin, ethanol has been shown to induce nervous system damage and affect the body in a variety of ways. Among the known effects of ethanol exposure are both transient and lasting consequences. Some of the lasting effects include long-term reduced neurogenesis in the hippocampus,[100][101] widespread brain atrophy,[102] and induced inflammation in the brain.[103] Of note, chronic ethanol ingestion has additionally been shown to induce reorganization of cellular membrane constituents, leading to a lipid bilayer marked by increased membrane concentrations of cholesterol and saturated fat.[50] This is important as neurotransmitter transport can be impaired through vesicular transport inhibition, resulting in diminished neural network function. One significant example of reduced inter-neuron communication is the ability for ethanol to inhibit NMDA receptors in the hippocampus, resulting in reduced long-term potentiation (LTP) and memory acquisition.[49] NMDA has been shown to play an important role in LTP and consequently memory formation.[104] With chronic ethanol intake, however, the susceptibility of these NMDA receptors to induce LTP increases in the mesolimbic dopamine neurons in an inositol 1,4,5-triphosphate (IP3) dependent manner.[105] This reorganization may lead to neuronal cytotoxicity both through hyperactivation of postsynaptic neurons and through induced addiction to continuous ethanol consumption. It has, additionally, been shown that ethanol directly reduces intracellular calcium ion accumulation through inhibited NMDA receptor activity, and thus reduces the capacity for the occurrence of LTP.[106]
In addition to the neurotoxic effects of ethanol in mature organisms, chronic ingestion is capable of inducing severe developmental defects. Evidence was first shown in 1973 of a connection between chronic ethanol intake by mothers and defects in their offspring.[107] This work was responsible for creating the classification of fetal alcohol syndrome, a disease characterized by common morphogenesis aberrations such as defects in craniofacial formation, limb development, and cardiovascular formation. The magnitude of ethanol neurotoxicity in fetuses leading to fetal alcohol syndrome has been shown to be dependent on antioxidant levels in the brain such as vitamin E.[108] As the fetal brain is relatively fragile and susceptible to induced stresses, severe deleterious effects of alcohol exposure can be seen in important areas such as the hippocampus and cerebellum. The severity of these effects is directly dependent upon the amount and frequency of ethanol consumption by the mother, and the stage in development of the fetus.[109] It is known that ethanol exposure results in reduced antioxidant levels, mitochondrial dysfunction (Chu 2007), and subsequent neuronal death, seemingly as a result of increased generation of reactive oxidative species (ROS).[30] This is a plausible mechanism, as there is a reduced presence in the fetal brain of antioxidant enzymes such as catalase and peroxidase.[110] In support of this mechanism, administration of high levels of dietary vitamin E results in reduced or eliminated ethanol-induced neurotoxic effects in fetuses.[8]
n-Hexane
n-Hexane is a neurotoxin which has been responsible for the poisoning of several workers in Chinese electronics factories in recent years.[111][112][113][51]
Receptor-selective neurotoxins
MPP+
MPP+, the toxic metabolite of MPTP is a selective neurotoxin which interferes with oxidative phosphorylation in mitochondria by inhibiting complex I, leading to the depletion of ATP and subsequent cell death. This occurs almost exclusively in dopaminergic neurons of the substantia nigra, resulting in the presentation of permanent parkinsonism in exposed subjects 2–3 days after administration.
Endogenous neurotoxin sources
Unlike most common sources of neurotoxins which are acquired by the body through ingestion, endogenous neurotoxins both originate from and exert their effects in-vivo. Additionally, though most venoms and exogenous neurotoxins will rarely possess useful in-vivo capabilities, endogenous neurotoxins are commonly used by the body in useful and healthy ways, such as nitric oxide which is used in cell communication.[114] It is often only when these endogenous compounds become highly concentrated that they lead to dangerous effects.[9]
Nitric oxide
Though nitric oxide (NO) is commonly used by the nervous system in inter-neuron communication and signaling, it can be active in mechanisms leading to ischemia in the cerebrum (Iadecola 1998). The neurotoxicity of NO is based on its importance in glutamate excitotoxicity, as NO is generated in a calcium-dependent manner in response to glutamate mediated NMDA activation, which occurs at an elevated rate in glutamate excitotoxicity.[52] Though NO facilitates increased blood flow to potentially ischemic regions of the brain, it is also capable of increasing oxidative stress,[115] inducing DNA damage and apoptosis.[116] Thus an increased presence of NO in an ischemic area of the CNS can produce significantly toxic effects.
Glutamate
Glutamate, like nitric oxide, is an endogenously produced compound used by neurons to perform normally, being present in small concentrations throughout the gray matter of the CNS.[9] One of the most notable uses of endogenous glutamate is its functionality as an excitatory neurotransmitter.[53] When concentrated, however, glutamate becomes toxic to surrounding neurons. This toxicity can be both a result of direct lethality of glutamate on neurons and a result of induced calcium flux into neurons leading to swelling and necrosis.[53] Support has been shown for these mechanisms playing significant roles in diseases and complications such as Huntington's disease, epilepsy, and stroke.[9]
See also
Notes
- ↑ Sivonen, K (1999). "Toxins produced by cyanobacteria". Vesitalous. 5: 11–18.
- ↑ Scottish Government Blue-Green Algae (Cyanobacteria) in Inland Waters: Assessment and Control of Risks to Public Health Retrieved 15 December 2011.
- ↑ Dorland's Medical Dictionary for Health Consumers
- 1 2 Spencer 2000
- 1 2 Olney 2002
- 1 2 3 4 5 6 7 8 Kiernan 2005
- ↑ Lidsky 2003
- 1 2 3 Heaton; Barrow, Marieta; Mitchell, J. Jean; Paiva, Michael (2000). "Amelioration of Ethanol-Induced Neurotoxicity in the Neonatal Rat Central Nervous System by Antioxidant Therapy". Alcoholism: Clinical and Experimental Research. 24 (4): 512–18. doi:10.1111/j.1530-0277.2000.tb02019.x. PMID 10798588.
- 1 2 3 4 Choi 1987
- ↑ Zhang 1994
- 1 2 Rosales, Raymond L.; Arimura, Kimiyoshi; Takenaga, Satoshi; Osame, Mitsuhiro (1996). "Extrafusal and Intrafusal Muscle Effects in Experimental Botulinum Toxin-A Injection". Muscle & Nerve. 19 (4): 488–96. doi:10.1002/(sici)1097-4598(199604)19:4<488::aid-mus9>3.0.co;2-8. PMID 8622728. S2CID 20849034.
- 1 2 Simpson 1986
- 1 2 3 Arnon 2001
- ↑ Dikranian 2001
- ↑ Deng 2003
- ↑ Jevtovic-Todorovic 2003
- ↑ Nadler 1978
- 1 2 3 4 Thyagarajan 2009
- ↑ Neurotoxins: Definition, Epidemiology, Etiology
- ↑ Hodge 2002
- 1 2 Dobbs 2009
- 1 2 3 Widmaier, Eric P., Hershel Raff, Kevin T. Strang, and Arthur J. Vander (2008) Vander's Human Physiology: the Mechanisms of Body Function.' Boston: McGraw-Hill Higher Education.
- 1 2 Martini 2009
- 1 2 Costa 2011
- ↑ Harry 1998
- ↑ Gartlon 2006
- ↑ Radio, Nicholas M.; Mundy, William R. (2008). "Developmental Neurotoxicity Testing in Vitro: Models for Assessing Chemical Effects on Neurite Out-growth". NeuroToxicology. 29 (3): 361–376. doi:10.1016/j.neuro.2008.02.011. PMID 18403021.
- ↑ Lotti 2005
- 1 2 3 Adams 2003
- 1 2 Brocardo 2011
- ↑ Lewendon 2001
- 1 2 Haghdoost-Yazdi 2011
- 1 2 DeBin 1993
- ↑ McClesky 1987
- 1 2 Garcia-Rodriguez 2011
- 1 2 Williamson 1996
- 1 2 Banks 1988
- 1 2 3 Aschner 1990
- 1 2 3 Dutertre 2006
- ↑ Koller 1988
- 1 2 Carmichael 1978
- 1 2 Carmichael 1975
- 1 2 Garcia-Cairasco, N.; Moyses-Neto, M.; Del Vecchio, F.; Oliveira, J. A. C.; Dos Santos, F. L.; Castro, O. W.; Arisi, G. M.; Dantas, M. R.; Carolino, R. O. G.; Coutinho-Netto, J.; Dagostin, A. L. A.; Rodrigues, M. C. A.; Leão, R. M.; Quintiliano, S. A. P.; Silva, L. F.; Gobbo-Neto, L.; Lopes, N. P. (2013). "Elucidating the Neurotoxicity of the Star Fruit". Angewandte Chemie International Edition. 52 (49): 13067–13070. doi:10.1002/anie.201305382. PMID 24281890.
- ↑ Rutgrere 2012
- ↑ Roller 1994
- 1 2 Konopacka 2009
- 1 2 DeFuria 2006
- 1 2 3 Bressler 1999
- 1 2 3 Lovinger, D.; White, G.; Weight, F. (1989). "Ethanol Inhibits NMDA-activated Ion Current in Hippocampal Neurons". Science. 243 (4899): 1721–724. Bibcode:1989Sci...243.1721L. doi:10.1126/science.2467382. PMID 2467382.
- 1 2 3 Leonard, B. E. (1986). "Is Ethanol a Neurotoxin?: the Effects of Ethanol on Neuronal Structure and Function". Alcohol and Alcoholism. 21 (4): 325–38. doi:10.1093/oxfordjournals.alcalc.a044638. PMID 2434114.
- 1 2 Occupational Safety and Health Guideline for n-Hexane Archived 2011-12-18 at the Wayback Machine, OSHA.gov
- 1 2 Garthwaite 1988
- 1 2 3 Choi 1990
- ↑ Ben-Shachar D, Zuk R, Glinka Y (1995). "Dopamine neurotoxicity: inhibition of mitochondrial respiration". J. Neurochem. 64 (2): 718–23. doi:10.1046/j.1471-4159.1995.64020718.x. PMID 7830065. S2CID 9060404.
- 1 2 3 Chowdhury, F. R.; Ahasan, H A M. Nazmul; Rashid, A K M. Mamunur; Mamun, A. Al; Khaliduzzaman, S. M. (2007). "Tetrodotoxin Poisoning: a Clinical Analysis, Role of Neostigmine and Short-term Outcome of 53 Cases". Singapore Medical Journal. 48 (9): 830–33. PMID 17728964.
- ↑ Ahasan 2004
- ↑ Lau 1995
- 1 2 3 Standfield 1983
- ↑ Roed 1989
- ↑ Haghdoost-Yasdi 2011
- ↑ Deshane 2003
- ↑ Soroceanu 1998
- 1 2 Jacob 2010
- ↑ Olivera 1987
- ↑ Cruz 1986
- ↑ McCleskey 1987
- 1 2 Brin, Mitchell F (1997) "Botulinum Toxin: Chemistry, Pharmacology, Toxicity, and Immunology." Muscle & Nerve, 20 (S6): 146–68.
- ↑ Montecucco 1986
- 1 2 Pirazzini 2011
- ↑ King 1981
- ↑ Rabe 1982
- ↑ Walton 2006
- ↑ Chan 2011
- ↑ Brookes 1988
- ↑ Yang 2007
- ↑ Wood 2007
- ↑ National Center for Environmental Assessment
- ↑ Devlin 1977
- ↑ Moore 1977
- ↑ Metcalf 2009
- ↑ Stewart 2008
- ↑ Dixit 2005
- ↑ Tsetlin 2003
- 1 2 Liu 2008
- ↑ Hue 2007
- 1 2 Bisset 1992
- ↑ Schlesinger 1946
- ↑ Griffith, Harold R.; Johnson, G. Enid (1942). "The Use Of Curare In General Anesthesia". Anesthesiology. 3 (4): 418–420. doi:10.1097/00000542-194207000-00006. S2CID 71400545.
- 1 2 Matsuoka 1991
- ↑ Buzanska (2000)
- 1 2 Norenberg 2004
- ↑ Liu 2009
- ↑ Vahidnia 2007
- 1 2 3 Rocha 2011
- ↑ Brender 2005
- ↑ DeFuria 2007
- 1 2 Lidskey 2003
- ↑ Bradbury 1993
- ↑ Lasley 1999
- ↑ Taffe 2010
- ↑ Morris 2009
- ↑ Bleich 2003
- ↑ Blanco 2005
- ↑ Davis 1992
- ↑ Bernier 2011
- ↑ Takadera 1990
- ↑ Jones 1973
- ↑ Mitchell 1999
- ↑ Gil-Mohapel 2010
- ↑ Bergamini 2004
- ↑ Workers poisoned while making iPhones ABC News, October 25, 2010
- ↑ Dirty Secrets Archived 2017-05-25 at the Wayback Machine ABC Foreign Correspondent, 2010-Oct-26
- ↑ Mr Daisey and the Apple Factory, This American Life, January 6, 2012
- ↑ Iadecola 1998
- ↑ Beckman 1990
- ↑ Bonfoco 1995
References
- Adams, Michael E.; Olivera, Baldomero M. (1994). "Neurotoxins: Overview of an Emerging Research Technology". Trends in Neurosciences. 17 (4): 151–55. doi:10.1016/0166-2236(94)90092-2. PMID 7517594. S2CID 3984106.
- Arnon, Stephen S.; Schechter, Robert; Inglesby, Thomas V.; Henderson, Donald A.; Bartlett, John G.; Ascher, Michael S.; Eitzen, Edward; Fine, Anne D.; Hauer, Jerome; Layton, Marcelle; Lillibridge, Scott; Osterholm, Michael T.; O'Toole, Tara; Parker, Gerald; Perl, Trish M.; Russell, Philip K.; Swerdlow, David L.; Tonat, Kevin (2001). "Botulinum Toxin as a Biological Weapon". The Journal of the American Medical Association. 285 (8): 1059–069. doi:10.1001/jama.285.8.1059. PMID 11209178.
- Aschner, M.; Aschner, J. (1990). "Mercury Neurotoxicity: Mechanisms of Blood–brain barrier Transport". Neuroscience & Biobehavioral Reviews. 14 (2): 169–76. doi:10.1016/s0149-7634(05)80217-9. PMID 2190116. S2CID 8777931.
- Banks, William A.; Kastin, Abba J. (1989). "Aluminum-Induced Neurotoxicity: Alterations in Membrane Function at the Blood–brain barrier". Neuroscience & Biobehavioral Reviews. 13 (1): 47–53. doi:10.1016/s0149-7634(89)80051-x. PMID 2671833. S2CID 46507895.
- Baum-Baicker, Cynthia (1985). "The Health Benefits of Moderate Alcohol Consumption: A Review of the Literature". Drug and Alcohol Dependence. 15 (3): 207–27. doi:10.1016/0376-8716(85)90001-8. PMID 4028954.
- Beckman, J. S. (1990). "Apparent Hydroxyl Radical Production by Peroxynitrite: Implications for Endothelial Injury from Nitric Oxide and Superoxide". Proceedings of the National Academy of Sciences. 87 (4): 1620–624. Bibcode:1990PNAS...87.1620B. doi:10.1073/pnas.87.4.1620. PMC 53527. PMID 2154753.
- Bergamini, Carlo M.; Gambetti, Stefani; Dondi, Alessia; Cervellati, Carlo (2004). "Oxygen, Reactive Oxygen Species and Tissue Damage". Current Pharmaceutical Design. 10 (14): 1611–626. doi:10.2174/1381612043384664. PMID 15134560.
- Bernier Brian E., Whitaker Leslie R., Morikawa Hitoshi (2011). "Previous Ethanol Experience Enhances Synaptic Plasticity of NMDA Receptors in the Ventral Tegmental Area". The Journal of Neuroscience. 31 (14): 5305–212. doi:10.1523/JNEUROSCI.5282-10.2011. PMC 3086894. PMID 21471355.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Bisset, Norman G (1992). "War and Hunting Poisons of the New World. Part 1. Notes on the Early History of Curare". Journal of Ethnopharmacology. 36 (1): 1–26. doi:10.1016/0378-8741(92)90056-w. PMID 1501489.
- Blanco, Ana M.; Valles, Soraya L.; Pascual, Maria; Guerri, Consuelo (2005). "Involvement of TLR4/Type I IL-1 Receptor Signaling in the Induction of Inflammatory Mediators and Cell Death Induced by Ethanol in Cultured Astrocytes". The Journal of Immunology. 175 (10): 6893–899. doi:10.4049/jimmunol.175.10.6893. PMID 16272348.
- Bleich, S (2003). "Hyperhomocysteinemia as a New Risk Factor for Brain Shrinkage in Patients with Alcoholism". Neuroscience Letters. 335 (3): 179–82. doi:10.1016/s0304-3940(02)01194-1. PMID 12531462. S2CID 33032529.
- Bonfoco E (1995). "Apoptosis and Necrosis: Two Distinct Events Induced, Respectively, by Mild and Intense Insults with N-Methyl-D-Aspartate or Nitric Oxide/Superoxide in Cortical Cell Cultures". Proceedings of the National Academy of Sciences. 92 (16): 7162–166. Bibcode:1995PNAS...92.7162B. doi:10.1073/pnas.92.16.7162. PMC 41299. PMID 7638161.
- Bradbury, MW; Deane, R (1993). "Permeability of the blood±brain barrier to lead. [Review]". Neurotoxicology. 14 (2–3): 131–6. PMID 8247388.
- Brender, J.; Suarez, L.; Felkner, M.; Gilani, Z.; Stinchcomb, D.; Moody, K.; Henry, J.; Hendricks, K. (2006). "Maternal Exposure to Arsenic, Cadmium, Lead, and Mercury and Neural Tube Defects in Offspring". Environmental Research. 101 (1): 132–39. Bibcode:2006ER....101..132B. doi:10.1016/j.envres.2005.08.003. PMID 16171797.
- Bressler, J; Kim, KA; Chakraborti, T; Goldstein, G (1999). "Molecular mechanisms of lead neurotoxicity. [Review]". Neurochem Res. 24 (4): 595–600. doi:10.1023/A:1022596115897. PMID 10227691. S2CID 3017703.
- Brocardo, Patricia S.; Gil-Mohapel, Joana; Christie, Brian R. (2011). "The Role of Oxidative Stress in Fetal Alcohol Spectrum Disorders". Brain Research Reviews. 67 (1–2): 209–25. doi:10.1016/j.brainresrev.2011.02.001. PMID 21315761. S2CID 1213492.
- Brookes, N (1988). "Specificity and Reversibility of the Inhibition by HgCl of Glutamate Transport in Astrocyte Cultures". Journal of Neurochemistry. 50 (4): 1117–122. doi:10.1111/j.1471-4159.1988.tb10581.x. PMID 2894409. S2CID 27522735.
- Buzanska, L.; Zablocka, B.; Dybel, A.; Domanska-Janik, K.; Albrecht, J. (2000). "Delayed Induction of Apoptosis by Ammonia in C6 Glioma Cells". Neurochemistry International. 37 (2–3): 287–97. doi:10.1016/s0197-0186(00)00030-9. PMID 10812214. S2CID 33351533.
- Carmichael WW, Biggs DF, Gorham PR (1975). "Toxicology and pharmacological action of Anabaena flos-aquae toxin". Science. 187 (4176): 542–544. Bibcode:1975Sci...187..542C. doi:10.1126/science.803708. PMID 803708.
- Carmichael WW, Gorham PR (1978). "Anatoxins from clones of Anabaena flos-aquae isolated from lakes of western Canada". Mitt. Infernal. Verein. Limnol. 21: 285–295.
- Chan, H. M. (2011) "Mercury in Fish: Human Health Risks." Encyclopedia of Environmental Health: 697–704.
- Choi, D (1988). "Calcium-mediated Neurotoxicity: Relationship to Specific Channel Types and Role in Ischemic Damage". Trends in Neurosciences. 11 (10): 465–69. doi:10.1016/0166-2236(88)90200-7. PMID 2469166. S2CID 36909560.
- Choi, D. W.; Rothman, S. M. (1990). "The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death". Annual Review of Neuroscience. 13 (1): 171–82. doi:10.1146/annurev.neuro.13.1.171. PMID 1970230.
- Choi, Dennis W (1987). "Ionic Dependence of Glutamate Neurotoxicity". The Journal of Neuroscience. 7 (2): 369–79. doi:10.1523/JNEUROSCI.07-02-00369.1987. PMC 6568907. PMID 2880938.
- Choi, Dennis W.; Maulucci-Gedde, Margaret; Kriegstein, Arnold R. (1987). "Glutamate Neurotoxicity in Cortical Cell Culture". The Journal of Neuroscience. 7 (2): 357–68. doi:10.1523/JNEUROSCI.07-02-00357.1987. PMC 6568898. PMID 2880937.
- Chu, Jennifer; Tong, Ming; Monte, Suzanne M. (2007). "Chronic Ethanol Exposure Causes Mitochondrial Dysfunction and Oxidative Stress in Immature Central Nervous System Neurons". Acta Neuropathologica. 113 (6): 659–73. doi:10.1007/s00401-007-0199-4. PMID 17431646. S2CID 25418731.
- Clancy, Barbara; Finlay, Barbara L.; Darlington, Richard B.; Anand, K.J.S. (2007). "Extrapolating Brain Development from Experimental Species to Humans". NeuroToxicology. 28 (5): 931–37. doi:10.1016/j.neuro.2007.01.014. PMC 2077812. PMID 17368774.
- Costa, Lucio G., Gennaro Giordano, and Marina Guizzetti (2011) In Vitro Neurotoxicology: Methods and Protocols. New York: Humana.
- Coyle, Joseph T.; Schwarcz, Robert (1976). "Lesion of Striatal Neurons with Kainic Acid Provides a Model for Huntington's Chorea". Nature. 246 (5574): 244–46. Bibcode:1976Natur.263..244C. doi:10.1038/263244a0. PMID 8731. S2CID 4271762.
- Cruz, Lourdes J.; Olivera, Baldomero M. (1987). "Calcium Channel Antagonists ω-Conotoxin Defines a New High Affinity Site". The Journal of Biological Chemistry. 14 (261): 6230–233.
- Davis, S.; Butcher, S. P.; Morris, R. (1992). "The NMDA Receptor Antagonist D-2-amino-5phosphonopentanoate (D-AP5) Impairs Spatial Learning and LTP in Vivo at Intracerebral Concentrations Comparable to Those That Block LTP in Vitro". The Journal of Neuroscience. 12 (1): 21–34. doi:10.1523/JNEUROSCI.12-01-00021.1992. PMC 6575679. PMID 1345945.
- Dawson, V. L. (1991). "Nitric Oxide Mediates Glutamate Neurotoxicity in Primary Cortical Cultures". Proceedings of the National Academy of Sciences. 88 (14): 6368–371. Bibcode:1991PNAS...88.6368D. doi:10.1073/pnas.88.14.6368. PMC 52084. PMID 1648740.
- Debin, John A., John E. Maggio, and Gary R. Strichartz (1993) "Purification and Characterization of Chlorotoxin, a Chloride Channel Ligand from the Venom of the Scorpion." The American Physiological Society, pp. 361–69.
- DeFuria, Jason; Shea, Thomas B. (2007). "Arsenic Inhibits Neurofilament Transport and Induces Perikaryal Accumulation of Phosphorylated Neurofilaments: Roles of JNK and GSK-3β". Brain Research. 1181: 74–82. doi:10.1016/j.brainres.2007.04.019. PMID 17961518. S2CID 37068398.
- Defuria, Jason (2006) "The Environmental Neurotoxin Arsenic Impairs Neurofilament Dynamics by Overactivation of C-JUN Terminal Kinase: Potential Role for Amyotrophic Lateral Sclerosis." UMI, pp. 1–16.
- Deng Wenbin, Poretz Ronald D (2003). "Oligodendroglia in Developmental Neurotoxicity". NeuroToxicology. 24 (2): 161–78. doi:10.1016/s0161-813x(02)00196-1. PMID 12606289.
- Deshane, Jessy; Garner, Craig C.; Sontheimer, Harald (2003). "Chlorotoxin Inhibits Glioma Cell Invasion via Matrix Metalloproteinase-2". The Journal of Biological Chemistry. 278 (6): 4135–144. doi:10.1074/jbc.m205662200. PMID 12454020.
- Devlin JP, Edwards OE, Gorham PR, Hunter NR, Pike RK, Stavric B (1977). "Anatoxin-a, a toxic alkaloid from Anabaena flos-aquae NRC-44h". Can. J. Chem. 55 (8): 1367–1371. doi:10.1139/v77-189.
- Dikranian, K (2001). "Apoptosis in the in Vivo Mammalian Forebrain". Neurobiology of Disease. 8 (3): 359–79. doi:10.1006/nbdi.2001.0411. PMID 11447994. S2CID 21965033.
- Dixit A, Dhaked RK, Alam SI, Singh L (2005). "Military potential of biological neurotoxins". Toxin Reviews. 24 (2): 175–207. doi:10.1081/TXR-200057850. S2CID 85651107.
- Dobbs, Michael R (2009) Clinical Neurotoxicology. Philadelphia: Saunders-Elsevier.
- Dutertre, S.; Lewis, R. (2006). "Toxin Insights into Nicotinic Acetylcholine Receptors". Biochemical Pharmacology. 72 (6): 661–70. doi:10.1016/j.bcp.2006.03.027. PMID 16716265.
- Garcia-Rodriguez, C.; Geren, I. N.; Lou, J.; Conrad, F.; Forsyth, C.; Wen, W.; Chakraborti, S.; Zao, H.; Manzanarez, G.; Smith, T. J.; Brown, J.; Tepp, W. H.; Liu, N.; Wijesuriya, S.; Tomic, M. T.; Johnson, E. A.; Smith, L. A.; Marks, J. D. (2011). "Response Re: 'Neutralizing Human Monoclonal Antibodies Binding Multiple Serotypes of Botulinum Neurotoxin' by Garcia-Rodriguez Et Al., PEDS, 2011;24:321–331". Protein Engineering Design and Selection. 24 (9): 633–34. doi:10.1093/protein/gzr012.
- Garthwaite, John; Charles, Sarah L.; Chess-Williams, Russel (1988). "Endothelim-derived Relaxing Factor Release on Activation of NMDA Receptors Suggests Role as Intercellular Messenger in the Brain". Nature. 336 (24): 385–88. Bibcode:1988Natur.336..385G. doi:10.1038/336385a0. PMID 2904125. S2CID 4361381.
- Gartlon, J.; Kinsner, A.; Balprice, A.; Coecke, S.; Clothier, R. (2006). "Evaluation of a Proposed in Vitro Test Strategy Using Neuronal and Non-neuronal Cell Systems for Detecting Neurotoxicity". Toxicology in Vitro. 20 (8): 1569–581. doi:10.1016/j.tiv.2006.07.009. PMID 16959468.
- Gil-Mohapel, Joana; Boehme, Fanny; Kainer, Leah; Christie, Brian R. (2010). "Hippocampal Cell Loss and Neurogenesis after Fetal Alcohol Exposure: Insights from Different Rodent Models". Brain Research Reviews. 64 (2): 283–303. doi:10.1016/j.brainresrev.2010.04.011. PMID 20471420. S2CID 10159395.
- Haghdoost-Yazdi, Hashem; Faraji, Ayda; Fraidouni, Negin; Movahedi, Mohadeseh; Hadibeygi, Elham; Vaezi, Fatemeh (2011). "Significant Effects of 4-aminopyridine and Tetraethylammonium in the Treatment of 6-hydroxydopamine-induced Parkinson's Disease". Behavioural Brain Research. 223 (1): 70–74. doi:10.1016/j.bbr.2011.04.021. PMID 21540059. S2CID 7100177.
- Harry, G. J.; Billingsley, Melvin; Bruinink, Arendd; Campbell, Iain L.; Classen, Werner; Dorman, David C.; Galli, Corrado; Ray, David; Smith, Robert A.; Tilson, Hugh A. (1998). "In Vitro Techniques for the Assessment of Neurotoxicity". Environmental Health Perspectives. 106 (Suppl 1): 131–58. doi:10.2307/3433917. JSTOR 3433917. PMC 1533280. PMID 9539010.
- Häussinger, Dieter (2006). "Low Grade Cerebral Edema and the Pathogenesis of Hepatic Encephalopathy in Cirrhosis". Hepatology. 43 (6): 1187–190. doi:10.1002/hep.21235. PMID 16729329.
- Hensley, K (1994). "A Model for β-Amyloid Aggregation and Neurotoxicity Based on Free Radical Generation by the Peptide: Relevance to Alzheimer Disease". Proceedings of the National Academy of Sciences. 91 (8): 3270–274. Bibcode:1994PNAS...91.3270H. doi:10.1073/pnas.91.8.3270. PMC 43558. PMID 8159737.
- Herbert, M. R. (2006) "Autism and Environmental Genomics." NeuroToxicology, pp. 671–84. Web.
- Hodge, A. Trevor (2002) Roman Aqueducts and Water Supply. London: Duckworth.
- How, C (2003). "Tetrodotoxin Poisoning". The American Journal of Emergency Medicine. 21 (1): 51–54. doi:10.1053/ajem.2003.50008. PMID 12563582.
- Hue, Bernard; Buckingham, Steven D.; Buckingham, David; Sattelle, David B. (2007). "Actions of Snake Neurotoxins on an Insect Nicotinic Cholinergic Synapse". Invertebrate Neuroscience. 7 (3): 173–78. doi:10.1007/s10158-007-0053-3. PMID 17710455. S2CID 31599093.
- Iadecola Constantino (1997). "Bright and Dark Sides of Nitric Oxide in Ischemic Brain Injury". Trends in Neurosciences. 20 (3): 132–39. doi:10.1016/s0166-2236(96)10074-6. PMID 9061868. S2CID 45011034.
- Jacob, Reed B.; McDougal, Owen M. (2010). "The M-superfamily of Conotoxins: a Review". Cellular and Molecular Life Sciences. 67 (1): 17–27. doi:10.1007/s00018-009-0125-0. PMC 3741454. PMID 19705062.
- Jevtovic-Todorovic, Vesna; Hartman, Richard E.; Izumi, Yukitoshi; Benshoff, Nicholas D.; Dikranian, Krikor; Zorumski, Charles F.; Olney, John W.; Wozniak, David F. (2003). "Early Exposure to Common Anesthetic Agents Causes Widespread Neurodegeneration in the Developing Rat Brain and Persistent Learning Deficits". The Journal of Neuroscience. 23 (3): 876–82. doi:10.1523/JNEUROSCI.23-03-00876.2003. PMC 6741934. PMID 12574416.
- Jones, K (1973). "Pattern Of Malformation In Offspring Of Chronic Alcoholic Mothers". The Lancet. 301 (7815): 1267–271. doi:10.1016/s0140-6736(73)91291-9. PMID 4126070.
- Kiernan, Matthew C.; Isbister, Geoffrey K.; Cindy; Lin, S.-Y.; Burke, David; Bostock, Hugh (2005). "Acute Tetrodotoxin-induced Neurotoxicity after Ingestion of Puffer Fish". Annals of Neurology. 57 (3): 339–48. doi:10.1002/ana.20395. PMID 15732107. S2CID 40455149.
- King, Steven W.; Savory, John; Wills, Michael R.; Gitelman, H. J. (1981). "The Clinical Biochemistry of Aluminum". Critical Reviews in Clinical Laboratory Sciences. 14 (1): 1–20. doi:10.3109/10408368109105861. PMID 7016437.
- Konopacka, Agnieszka; Konopacki, Filip A.; Albrecht, Jan (2009). "Protein Kinase G Is Involved in Ammonia-induced Swelling of Astrocytes". Journal of Neurochemistry. 109: 246–51. doi:10.1111/j.1471-4159.2009.05802.x. PMID 19393034. S2CID 42719321.
- Lafon-Cazal, Mireille; Pietri, Sylvia; Culcasi, Marcel; Bockaert, Joel (1993). "NMDA-dependent Superoxide Production and Neurotoxicity". Nature. 364 (6437): 535–37. Bibcode:1993Natur.364..535L. doi:10.1038/364535a0. PMID 7687749. S2CID 4246917.
- Lasley, SM; Green, MC; Gilbert, ME (1999). "Influence of exposure period on in vivo hippocampal glutamate and GABA release in rats chronically exposed to lead". Neurotoxicology. 20 (4): 619–29. PMID 10499360.
- Lau, F. L.; Wong, C. K.; Yip, S. H. (1995). "Puffer Fish Poisoning". Emergency Medicine Journal. 12 (3): 214–15. doi:10.1136/emj.12.3.214. PMC 1342486. PMID 8581253.
- Lewendon, G.; Kinra, S.; Nelder, R.; Cronin, T. (2001). "Should Children with Developmental and Behavioural Problems Be Routinely Screened for Lead?". Archives of Disease in Childhood. 85 (4): 286–88. doi:10.1136/adc.85.4.286. PMC 1718950. PMID 11567935.
- Lidsky Theodore I (2003). "Lead Neurotoxicity in Children: Basic Mechanisms and Clinical Correlates". Brain. 126 (1): 5–19. doi:10.1093/brain/awg014. PMID 12477693.
- Liu, Kuang-Kai; Chen, Mei-Fang; Chen, Po-Yi; Lee, Tony J F.; Cheng, Chia-Liang; Chang, Chia-Ching; Ho, Yen-Peng; Jui-I, Chao (2008). "Alpha-bungarotoxin Binding to Target Cell in a Developing Visual System by Carboxylated Nanodiamond". Nanotechnology. 19 (20): 205102. Bibcode:2008Nanot..19t5102L. doi:10.1088/0957-4484/19/20/205102. PMID 21825732. S2CID 1260143.
- Liu, Yuan; McDermott, Suzanne; Lawson, Andrew; Aelion, C. Marjorie (2010). "The Relationship between Mental Retardation and Developmental Delays in Children and the Levels of Arsenic, Mercury and Lead in Soil Samples Taken near Their Mother's Residence during Pregnancy". International Journal of Hygiene and Environmental Health. 213 (2): 116–23. doi:10.1016/j.ijheh.2009.12.004. PMC 2836425. PMID 20045663.
- Lorenzo, A (1994). "β-Amyloid Neurotoxicity Requires Fibril Formation and Is Inhibited by Congo Red". Proceedings of the National Academy of Sciences. 91 (25): 12243–2247. Bibcode:1994PNAS...9112243L. doi:10.1073/pnas.91.25.12243. PMC 45413. PMID 7991613.
- Lotti, Marcello, and Angelo Moretto (1989) "Organophosphate-Induced Delayed Polyneuropathy." Toxicological Reviews, 24 (1) (2005): 37–49.
- Martinez-Finley, Ebany J.; Goggin, Samantha L.; Labrecque, Matthew T.; Allan, Andrea M. (2011). "Reduced Expression of MAPK/ERK Genes in Perinatal Arsenic-exposed Offspring Induced by Glucocorticoid Receptor Deficits". Neurotoxicology and Teratology. 33 (5): 530–37. doi:10.1016/j.ntt.2011.07.003. PMC 3183307. PMID 21784148.
- Martini, Frederic, Michael J. Timmons, and Robert B. Tallitsch (2009) Human Anatomy. San Francisco: Pearson/Benjamin Cummings.
- Matsuoka, Masato; Igisu, Hideki; Kohriyama, Kazuaki; Inoue, Naohide (1991). "Suppression of Neurotoxicity of Ammonia by L-carnitine". Brain Research. 567 (2): 328–31. doi:10.1016/0006-8993(91)90814-c. PMID 1817738. S2CID 10250413.
- McCleskey, E. W. (1987). "Omega-conotoxin: Direct and Persistent Blockade of Specific Types of Calcium Channels in Neurons but Not Muscle". Proceedings of the National Academy of Sciences. 84 (12): 4327–331. Bibcode:1987PNAS...84.4327M. doi:10.1073/pnas.84.12.4327. PMC 305078. PMID 2438698.
- Meldrum, B.; Garthwaite, J. (1990). "Excitatory Amino Acid Neurotoxicity and Neurodegenerative Disease". Trends in Pharmacological Sciences. 11 (9): 379–87. doi:10.1016/0165-6147(90)90184-a. PMID 2238094.
- Metcalf, James S.; Codd, Geoffrey A. (2009). "Cyanobacteria, neurotoxins and water resources: Are there implications for human neurodegenerative disease?". Amyotrophic Lateral Sclerosis. 10: 74–78. doi:10.3109/17482960903272942. PMID 19929737. S2CID 41880444.
- Mitchell, J.Jean; Paiva, Michael; Barrow Heaton, Marieta (1999). "The Antioxidants Vitamin E and β-Carotene Protect Against Ethanol-Induced Neurotoxicity in Embryonic Rat Hippocampal Cultures". Alcohol. 17 (2): 163–68. doi:10.1016/s0741-8329(98)00051-2. PMID 10064385.
- Moore RE (1977). "Toxins from blue-green algae". BioScience. 27 (12): 797–802. doi:10.2307/1297756. JSTOR 1297756.
- Montecucco C (1986). "How Do Tetanus and Botulinum Toxins Bind to Neuronal Membranes?". Trends in Biochemical Sciences. 11 (8): 314–17. doi:10.1016/0968-0004(86)90282-3.
- Morris, Stephanie A., David W. Eaves, Aleksander R. Smith, and Kimberly Nixon (2009) "Alcohol Inhibition of Neurogenesis: A Mechanism of Hippocampal Neurodegeneration in an Adolescent Alcohol Abuse Model." Hippocampus: NA.
- Nadler; Victor, J.; Perry, Bruce W.; Cotman, Carl W. (1978). "Intraventricular Kainic Acid Preferentially Destroys Hippocampal Pyramidal Cells". Nature. 271 (5646): 676–77. Bibcode:1978Natur.271..676N. doi:10.1038/271676a0. PMID 625338. S2CID 4267990.
- National Center for Environmental Assessment (2006) "Toxicological Reviews of Cyanobacterial Toxins: Anatoxin-a" NCEA-C-1743
- Norenberg, M. D.; Rao, K. V. Rama; Jayakumar, A. R. (2004). "Ammonia Neurotoxicity and the Mitochondrial Permeability Transition". Journal of Bioenergetics and Biomembranes. 36 (4): 303–07. doi:10.1023/b:jobb.0000041758.20071.19. PMID 15377862. S2CID 39221153.
- Olivera, Baldomero M.; Cruz, Lourdes J.; De Santos, Victoria; LeCheminant, Garth; Griffin, David; Zeikus, Regina; McIntosh, J. Michael; Galyean, Robert; Varga, Janos (1987). "Neuronal Calcium Channel Antagonists. Discrimination between Calcium Channel Subtypes Using .omega.-conotoxin from Conus Magus Venom". Biochemistry. 26 (8): 2086–090. doi:10.1021/bi00382a004. PMID 2441741.
- Olney, John W (2002). "New Insights and New Issues in Developmental Neurotoxicology". NeuroToxicology. 23 (6): 659–68. doi:10.1016/s0161-813x(01)00092-4. PMID 12520755.
- Pirazzini, Marco, Ornella Rossetto, Paolo Bolognese, Clifford C. Shone, and Cesare Montecucco (2011) "Double Anchorage to the Membrane and Intact Inter-chain Disulfide Bond Are Required for the Low PH Induced Entry of Tetanus and Botulinum Neurotoxins into Neurons." Cellular Microbiology: No. Print.
- Rabe, Ausma; He Lee, Moon; Shek, Judy; Wisniewski, Henryk M. (1982). "Learning Deficit in Immature Rabbits with Aluminum-induced Neurofibrillary Changes". Experimental Neurology. 76 (2): 441–46. doi:10.1016/0014-4886(82)90220-5. PMID 6896495. S2CID 7495252.
- Rocha, R. A.; Gimeno-Alcaniz, J. V.; Martín-Ibanez, Raymond; Canals, J. M.; Vélez, D.; Devesa, V. (2011). "Arsenic and Fluoride Induce Neural Progenitor Cell Apoptosis". Toxicology Letters. 203 (3): 237–44. doi:10.1016/j.toxlet.2011.03.023. PMID 21439358.
- Roed, A (1989). "The Effects of Tetraethylammonium during Twitch and Tetanic Stimulation of the Phrenic Nerve Diaphragm Preparation in the Rat". Neuropharmacology. 28 (6): 585–92. doi:10.1016/0028-3908(89)90137-8. PMID 2547180. S2CID 1916154.
- Rothman, S.; Thurston, J.; Hauhart, R. (1987). "Delayed Neurotoxicity of Excitatory Amino Acids In Vitro". Neuroscience. 22 (2): 471–80. doi:10.1016/0306-4522(87)90347-2. PMID 3670595. S2CID 6038419.
- Schlesinger, Edward B (1946). "Curare A Review of Its Therapeutic Effects and Their Physiological Basis". The American Journal of Medicine. 1 (5): 518–30. doi:10.1016/0002-9343(46)90073-3. PMID 21001466.
- Siebler, M.; Koller, H.; Schmalenbach, C.; Muller, H. (1988). "GABA Activated Chloride Currents in Cultured Rat Hippocampal and Septal Region Neurons Can Be Inhibited by Curare and Atropine". Neuroscience Letters. 93 (2–3): 220–24. doi:10.1016/0304-3940(88)90085-7. PMID 2468110. S2CID 10645685.
- Simpson, L. L. (1986). "Molecular Pharmacology of Botulinum Toxin and Tetanus Toxin". Annual Review of Pharmacology and Toxicology. 26 (1): 427–53. doi:10.1146/annurev.pharmtox.26.1.427. PMID 3521461.
- Soroceanu, Liliana; Gillespie, Yancey; Khazaeli, M. B.; Sontheimer, Harold (1998). "Use of Chlorotoxin for Targeting of Primary Brain Tumors". Cancer Research. 58 (21): 4871–879. PMID 9809993.
- Spencer PS, Schaumburg HH, Ludolph AC (Eds) (2000) Experimental and Clinical Neurotoxicology. Oxford University Press, Oxford, pp. 1310.
- Stanfield, Peter R (1983). "Tetraethylammonium Ions and the Potassium Permeability of Excitable Cells". Reviews of Physiology, Biochemistry and Pharmacology. 97: 1–49. doi:10.1007/bfb0035345. ISBN 978-3-540-11701-8. PMID 6306751.
- Stewart I, Seawright AA, Shaw GR (2008). "Cyanobacterial poisoning in livestock, wild mammals and birds – an overview" (PDF). Cyanobacterial Harmful Algal Blooms: State of the Science and Research Needs. Advances in Experimental Medicine and Biology. Vol. 619. pp. 613–637. doi:10.1007/978-0-387-75865-7_28. ISBN 978-0-387-75864-0. PMID 18461786.
- Taffe, M. A.; Kotzebue, R. W.; Crean, R. D.; Crawford, E. F.; Edwards, S.; Mandyam, C. D. (2010). "From the Cover: Long-lasting Reduction in Hippocampal Neurogenesis by Alcohol Consumption in Adolescent Nonhuman Primates". Proceedings of the National Academy of Sciences. 107 (24): 11104–1109. Bibcode:2010PNAS..10711104T. doi:10.1073/pnas.0912810107. PMC 2890755. PMID 20534463.
- Takadera, Tsuneo; Suzuki, Risa; Mohri, Tetsuro (1990). "Protection by Ethanol of Cortical Neurons from N-methyl-d-aspartate-induced Neurotoxicity Is Associated with Blocking Calcium Influx". Brain Research. 537 (1–2): 109–14. doi:10.1016/0006-8993(90)90346-d. PMID 1982237. S2CID 22160700.
- Thyagarajan, B.; Krivitskaya, N.; Potian, J. G.; Hognason, K.; Garcia, C. C.; McArdle, J. J. (2009). "Capsaicin Protects Mouse Neuromuscular Junctions from the Neuroparalytic Effects of Botulinum Neurotoxin A." Journal of Pharmacology and Experimental Therapeutics. 331 (2): 361–71. doi:10.1124/jpet.109.156901. PMC 2775269. PMID 19654265.
- Tymianski, Michael; Charlton, Milton P.; Carlen, Peter L.; Tator, Charles H. (2003). "Source Specificity of Early Calcium Neurotoxicity in Cultured Embryonic Spinal Neurons". The Journal of Neuroscience. 13 (5): 2095–104. doi:10.1523/JNEUROSCI.13-05-02085.1993. PMC 6576557. PMID 8097530.
- Tsetlin, V.I; Hucho, F. (2004). "Snake and Snail Toxins Acting on Nicotinic Acetylcholine Receptors: Fundamental Aspects and Medical Applications". FEBS Letters. 557 (1–3): 9–13. doi:10.1016/s0014-5793(03)01454-6. PMID 14741333.
- USEPA (United States Environmental Protection Agency) (1998) Health Effects Test Guidelines. OPPTS 870.6200. Neurotoxicity screening battery. Washington DC, USEPA.
- Vahidnia, A., G.B. Van Der Voet, and F.A. De Wolff (2007) "Arsenic Neurotoxicity A Review." Human & Experimental Toxicology, 26 (10) : 823–32.
- Walton, J (2006). "Aluminum in Hippocampal Neurons from Humans with Alzheimer's Disease". NeuroToxicology. 27 (3): 385–94. doi:10.1016/j.neuro.2005.11.007. PMID 16458972.
- Widmaier, Eric P., Hershel Raff, Kevin T. Strang, and Arthur J. Vander (2008) Vander's Human Physiology: the Mechanisms of Body Function. Boston: McGraw-Hill Higher Education.
- Williamson, Lura C.; Halpern, Jane L.; Montecucco, Cesare; Brown, J. E.; Neale, Elaine A. (1996). "Clostridial Neurotoxins and Substrate Proteolysis in Intact Neurons". The Journal of Biological Chemistry. 271 (13): 7694–699. doi:10.1074/jbc.271.13.7694. PMID 8631808.
- Wood S. A.; Rasmussen J. P.; Holland P. T.; Campbell R.; Crowe A. L. M. (2007). "First Report of the Cyanotoxin Anatoxin-A from Aphanizomenon issatschenkoi (cyanobacteria)". Journal of Phycology. 43 (2): 356–365. doi:10.1111/j.1529-8817.2007.00318.x. S2CID 84284928.
- Yamada, Kiyofumi; Tanaka, Tomoko; Han, Daiken; Senzaki, Kouji; Kameyama, Tsutomu; Nabeshima, Toshitaka (1999). "Protective Effects of Idebenone and α-tocopherol on β-amyloid-(1–42)-induced Learning and Memory Deficits in Rats: Implication of Oxidative Stress in β-amyloid-induced Neurotoxicity In vivo". European Journal of Neuroscience. 11 (1): 83–90. doi:10.1046/j.1460-9568.1999.00408.x. PMID 9987013. S2CID 84408095.
- Yan; Du, Shi; Chen, Xi; Fu, Jin; Chen, Ming; Zhu, Huaijie; Roher, Alex; Slattery, Timothy; Zhao, Lei; Nagashima, Mariko; Morser, John; Migheli, Antonio; Nawroth, Peter; Stern, David; Marie Schmidt, Ann (1996). "RAGE and Amyloid-β Peptide Neurotoxicity in Alzheimer's Disease". Nature. 382 (6593): 685–91. Bibcode:1996Natur.382..685Y. doi:10.1038/382685a0. PMID 8751438. S2CID 4372023.
- Yang, X (2007) Occurrence of the cyanobacterial neurotoxin, anatoxin-a, in New York State waters ProQuest. ISBN 978-0-549-35451-2.
- Zhang, J.; Dawson, V.; Dawson, T.; Snyder, S. (1994). "Nitric Oxide Activation of Poly(ADP-ribose) Synthetase in Neurotoxicity". Science. 263 (5147): 687–89. Bibcode:1994Sci...263..687Z. doi:10.1126/science.8080500. PMID 8080500.
Further reading
- Brain Facts Book at The Society for Neuroscience
- Neuroscience Texts at University of Texas Medical School
- In Vitro Neurotoxicology: An Introduction at Springerlink
- Biology of the NMDA Receptor at NCBI
- Advances in the Neuroscience of Addiction, 2nd edition at NCBI
External links
- Environmental Protection Agency at United States Environmental Protection Agency
- Alcohol and Alcoholism at Oxford Medical Journals
- Neurotoxicology at Elsevier Journals
- Neurotoxin Institute at Neurotoxin Institute
- Neurotoxins] at Toxipedia
