A neutral network is a set of genes all related by point mutations that have equivalent function or fitness.[1] Each node represents a gene sequence and each line represents the mutation connecting two sequences. Neutral networks can be thought of as high, flat plateaus in a fitness landscape. During neutral evolution, genes can randomly move through neutral networks and traverse regions of sequence space which may have consequences for robustness and evolvability.
Genetic and molecular causes
Neutral networks exist in fitness landscapes since proteins are robust to mutations. This leads to extended networks of genes of equivalent function, linked by neutral mutations.[2][3] Proteins are resistant to mutations because many sequences can fold into highly similar structural folds.[4] A protein adopts a limited ensemble of native conformations because those conformers have lower energy than unfolded and mis-folded states (ΔΔG of folding).[5][6] This is achieved by a distributed, internal network of cooperative interactions (hydrophobic, polar and covalent).[7] Protein structural robustness results from few single mutations being sufficiently disruptive to compromise function. Proteins have also evolved to avoid aggregation[8] as partially folded proteins can combine to form large, repeating, insoluble protein fibrils and masses.[9] There is evidence that proteins show negative design features to reduce the exposure of aggregation-prone beta-sheet motifs in their structures.[10] Additionally, there is some evidence that the genetic code itself may be optimised such that most point mutations lead to similar amino acids (conservative).[11][12] Together these factors create a distribution of fitness effects of mutations that contains a high proportion of neutral and nearly-neutral mutations.[13]
Evolution
Neutral networks are a subset of the sequences in sequence space that have equivalent function, and so form a wide, flat plateau in a fitness landscape. Neutral evolution can therefore be visualised as a population diffusing from one set of sequence nodes, through the neutral network, to another cluster of sequence nodes. Since the majority of evolution is thought to be neutral,[14][15] a large proportion of gene change is the movement though expansive neutral networks.
Robustness
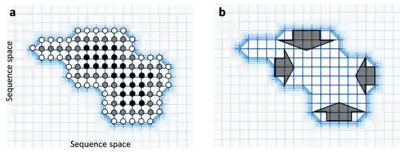
The more neutral neighbours a sequence has, the more robust to mutations it is since mutations are more likely to simply neutrally convert it into an equally functional sequence.[1] Indeed, if there are large differences between the number of neutral neighbours of different sequences within a neutral network, the population is predicted to evolve towards these robust sequences. This is sometimes called circum-neutrality and represents the movement of populations away from cliffs in the fitness landscape.[16]
In addition to in silico models,[17] these processes are beginning to be confirmed by experimental evolution of cytochrome P450s[18] and B-lactamase.[19]
Evolvability
Interest in the interplay between genetic drift and selection has been around since the 1930s when the shifting-balance theory proposed that in some situations, genetic drift could facilitate later adaptive evolution.[20] Although the specifics of the theory were largely discredited,[21] it drew attention to the possibility that drift could generate cryptic variation that, though neutral to current function, may affect selection for new functions (evolvability).[22]
By definition, all genes in a neutral network have equivalent function, however some may exhibit promiscuous activities which could serve as starting points for adaptive evolution towards new functions.[23][24] In terms of sequence space, current theories predict that if the neutral networks for two different activities overlap, a neutrally evolving population may diffuse to regions of the neutral network of the first activity that allow it to access the second.[25] This would only be the case when the distance between activities is smaller than the distance that a neutrally evolving population can cover. The degree of interpenetration of the two networks will determine how common cryptic variation for the promiscuous activity is in sequence space.[26]
Mathematical Framework
The fact that neutral mutations were probably widespread was proposed by Freese and Yoshida in 1965.[27] Motoo Kimura later crystallized a theory of neutral evolution in 1968[28] with King and Jukes independently proposing a similar theory (1969).[29] Kimura computed the rate of nucleotide substitutions in a population (i.e. the average time for one base pair replacement to occur within a genome) and found it to be ~1.8 years. Such a high rate would not be tolerated by any mammalian population according to Haldane's formula. He thus concluded that, in mammals, neutral (or nearly neutral) nucleotide substitution mutations of DNA must dominate. He computed that such mutations were occurring at the rate of roughly 0-5 per year per gamete.
In later years, a new paradigm emerged, that placed RNA as a precursor molecule to DNA. A primordial molecule principle was put forth as early as 1968 by Crick,[30] and lead to what is now known as The RNA World Hypothesis.[31] DNA is found, predominantly, as fully base paired double helices, while biological RNA is single stranded and often exhibits complex base-pairing interactions. These are due to its increased ability to form hydrogen bonds, a fact which stems from the existence of the extra hydroxyl group in the ribose sugar.
In the 1970s, Stein and M. Waterman laid the groundwork for the combinatorics of RNA secondary structures.[32] Waterman gave the first graph theoretic description of RNA secondary structures and their associated properties, and used them to produce an efficient minimum free energy (MFE) folding algorithm.[33] An RNA secondary structure can be viewed as a diagram over N labeled vertices with its Watson-Crick base pairs represented as non-crossing arcs in the upper half plane. Therefore, a secondary structure is a scaffold having many sequences compatible with its implied base pairing constraints. Later, Smith and Waterman developed an algorithm that performed local sequence alignment.[34] Another prediction algorithm for RNA secondary structure was given by Nussinov[35] Nussinov's algorithm described the folding problem over a two letter alphabet as a planar graph optimization problem, where the quantity to be maximized is the number of matchings in the sequence string.
Come the year 1980, Howell et al. computed a generating function of all foldings of a sequence[36] while D. Sankoff (1985) described algorithms for alignment of finite sequences, the prediction of RNA secondary structures (folding), and the reconstruction of proto-sequences on a phylo-genetic tree.[37] Later, Waterman and Temple (1986) produced a polynomial time dynamic programming (DP) algorithm for predicting general RNA secondary structure.[38] while in the year 1990, John McCaskill presented a polynomial time DP algorithm for computing the full equilibrium partition function of an RNA secondary structure.[39]
M. Zuker, implemented algorithms for computation of MFE RNA secondary structures[40] based on the work of Nussinov et al.,[35] Smith and Waterman[34] and Studnicka, et al.[41] Later L. Hofacker (et al., 1994),[42] presented The Vienna RNA package, a software package that integrated MFE folding and the computation of the partition function as well as base pairing probabilities.
Peter Schuster and W. Fontana (1994) shifted the focus towards sequence to structure maps (genotype–phenotype) . They used an inverse folding algorithm, to produce computational evidence that RNA sequences sharing the same structure are distributed randomly in sequence space. They observed that common structures can be reached from a random sequence by just a few mutations. These two facts lead them to conclude that the sequence space seemed to be percolated by neutral networks of nearest neighbor mutants that fold to the same structure.[43]
In 1997, C. Reidys Stadler and Schuster laid the mathematical foundations for the study and modelling of neutral networks of RNA secondary structures. Using a random graph model they proved the existence of a threshold value for connectivity of random sub-graphs in a configuration space, parametrized by λ, the fraction of neutral neighbors. They showed that the networks are connected and percolate sequence space if the fraction of neutral nearest neighbors exceeds λ*, a threshold value. Below this threshold the networks are partitioned into a largest giant component and several smaller ones. Key results of this analysis where concerned with threshold functions for density and connectivity for neutral networks as well as Schuster's shape space conjecture.[43][44][45]
See also
References
- 1 2 van Nimwegen, E; Crutchfield, JP; Huynen, M (Aug 17, 1999). "Neutral evolution of mutational robustness". Proceedings of the National Academy of Sciences of the United States of America. 96 (17): 9716–20. arXiv:adap-org/9903006. Bibcode:1999PNAS...96.9716V. doi:10.1073/pnas.96.17.9716. PMC 22276. PMID 10449760.
- ↑ Taverna, DM; Goldstein, RA (Jan 18, 2002). "Why are proteins so robust to site mutations?". Journal of Molecular Biology. 315 (3): 479–84. doi:10.1006/jmbi.2001.5226. PMID 11786027.
- ↑ Tokuriki, N; Tawfik, DS (Oct 2009). "Stability effects of mutations and protein evolvability". Current Opinion in Structural Biology. 19 (5): 596–604. doi:10.1016/j.sbi.2009.08.003. PMID 19765975.
- ↑ Meyerguz, L; Kleinberg, J; Elber, R (Jul 10, 2007). "The network of sequence flow between protein structures". Proceedings of the National Academy of Sciences of the United States of America. 104 (28): 11627–32. Bibcode:2007PNAS..10411627M. doi:10.1073/pnas.0701393104. PMC 1913895. PMID 17596339.
- ↑ Karplus, M (Jun 17, 2011). "Behind the folding funnel diagram". Nature Chemical Biology. 7 (7): 401–4. doi:10.1038/nchembio.565. PMID 21685880.
- ↑ Tokuriki, N; Stricher, F; Schymkowitz, J; Serrano, L; Tawfik, DS (Jun 22, 2007). "The stability effects of protein mutations appear to be universally distributed". Journal of Molecular Biology. 369 (5): 1318–32. doi:10.1016/j.jmb.2007.03.069. PMID 17482644. S2CID 24638570.
- ↑ Shakhnovich, BE; Deeds, E; Delisi, C; Shakhnovich, E (Mar 2005). "Protein structure and evolutionary history determine sequence space topology". Genome Research. 15 (3): 385–92. arXiv:q-bio/0404040. doi:10.1101/gr.3133605. PMC 551565. PMID 15741509.
- ↑ Monsellier, E; Chiti, F (Aug 2007). "Prevention of amyloid-like aggregation as a driving force of protein evolution". EMBO Reports. 8 (8): 737–42. doi:10.1038/sj.embor.7401034. PMC 1978086. PMID 17668004.
- ↑ Fink, AL (1998). "Protein aggregation: folding aggregates, inclusion bodies and amyloid". Folding & Design. 3 (1): R9-23. doi:10.1016/s1359-0278(98)00002-9. PMID 9502314.
- ↑ Richardson, JS; Richardson, DC (Mar 5, 2002). "Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation". Proceedings of the National Academy of Sciences of the United States of America. 99 (5): 2754–9. Bibcode:2002PNAS...99.2754R. doi:10.1073/pnas.052706099. PMC 122420. PMID 11880627.
- ↑ Müller, MM; Allison, JR; Hongdilokkul, N; Gaillon, L; Kast, P; van Gunsteren, WF; Marlière, P; Hilvert, D (2013). "Directed evolution of a model primordial enzyme provides insights into the development of the genetic code". PLOS Genetics. 9 (1): e1003187. doi:10.1371/journal.pgen.1003187. PMC 3536711. PMID 23300488.
- ↑ Firnberg, E; Ostermeier, M (Aug 2013). "The genetic code constrains yet facilitates Darwinian evolution". Nucleic Acids Research. 41 (15): 7420–8. doi:10.1093/nar/gkt536. PMC 3753648. PMID 23754851.
- ↑ Hietpas, RT; Jensen, JD; Bolon, DN (May 10, 2011). "Experimental illumination of a fitness landscape". Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. Bibcode:2011PNAS..108.7896H. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- ↑ Kimura, Motoo. (1983). The neutral theory of molecular evolution. Cambridge
- ↑ Kimura, M. (1968). "Evolutionary Rate at the Molecular Level". Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- ↑ Proulx, SR; Adler, FR (Jul 2010). "The standard of neutrality: still flapping in the breeze?". Journal of Evolutionary Biology. 23 (7): 1339–50. doi:10.1111/j.1420-9101.2010.02006.x. PMID 20492093. S2CID 7774510.
- ↑ van Nimwegen E.; Crutchfield J. P.; Huynen M. (1999). "Neutral evolution of mutational robustness". PNAS. 96 (17): 9716–9720. Bibcode:1999PNAS...96.9716V. doi:10.1073/pnas.96.17.9716. PMC 22276. PMID 10449760.
- ↑ Bloom, JD; Lu, Z; Chen, D; Raval, A; Venturelli, OS; Arnold, FH (Jul 17, 2007). "Evolution favors protein mutational robustness in sufficiently large populations". BMC Biology. 5: 29. arXiv:0704.1885. doi:10.1186/1741-7007-5-29. PMC 1995189. PMID 17640347.
- ↑ Bershtein, Shimon; Goldin, Korina; Tawfik, Dan S. (June 2008). "Intense Neutral Drifts Yield Robust and Evolvable Consensus Proteins". Journal of Molecular Biology. 379 (5): 1029–1044. doi:10.1016/j.jmb.2008.04.024. PMID 18495157.
- ↑ Wright, Sewel (1932). "The roles of mutation, inbreeding, crossbreeding and selection in evolution". Proceedings of the Sixth International Congress of Genetics: 356–366.
- ↑ Coyne, JA; Barton NH; Turelli M (1997). "Perspective: a critique of Sewall Wright's shifting balance theory of evolution". Evolution. 51 (3): 643–671. doi:10.2307/2411143. JSTOR 2411143. PMID 28568586.
- ↑ Davies, E. K. (10 September 1999). "High Frequency of Cryptic Deleterious Mutations in Caenorhabditis elegans". Science. 285 (5434): 1748–1751. doi:10.1126/science.285.5434.1748. PMID 10481013.
- ↑ Masel, J (Mar 2006). "Cryptic genetic variation is enriched for potential adaptations". Genetics. 172 (3): 1985–91. doi:10.1534/genetics.105.051649. PMC 1456269. PMID 16387877.
- ↑ Hayden, EJ; Ferrada, E; Wagner, A (Jun 2, 2011). "Cryptic genetic variation promotes rapid evolutionary adaptation in an RNA enzyme" (PDF). Nature. 474 (7349): 92–5. doi:10.1038/nature10083. PMID 21637259. S2CID 4390213.
- ↑ Bornberg-Bauer, E; Huylmans, AK; Sikosek, T (Jun 2010). "How do new proteins arise?". Current Opinion in Structural Biology. 20 (3): 390–6. doi:10.1016/j.sbi.2010.02.005. PMID 20347587.
- ↑ Wagner, Andreas (2011-07-14). The origins of evolutionary innovations : a theory of transformative change in living systems. Oxford [etc.]: Oxford University Press. ISBN 978-0199692590.
- ↑ Freese, E. and Yoshida, A. (1965). The role of mutations in evolution. In V Bryson, and H J Vogel, eds. Evolving Genes and Proteins, pp. 341-55. Academic, New York.
- ↑ Kimura, M (1968). "Evolutionary Rate at the Molecular Level". Nature. 217 (5129): 624–6. Bibcode:1968Natur.217..624K. doi:10.1038/217624a0. PMID 5637732. S2CID 4161261.
- ↑ King, JL; Jukes, TH (1969). "Non-Darwinian Evolution". Science. 164 (3881): 788–97. Bibcode:1969Sci...164..788L. doi:10.1126/science.164.3881.788. PMID 5767777.
- ↑ Crick, FH (1968). "The origin of the genetic code". Journal of Molecular Biology. 38 (3): 367–79. doi:10.1016/0022-2836(68)90392-6. PMID 4887876.
- ↑ Robertson, MP; Joyce, GF (2012). "The origins of the RNA world". Cold Spring Harbor Perspectives in Biology. 4 (5): a003608. doi:10.1101/cshperspect.a003608. PMC 3331698. PMID 20739415.
- ↑ Stein, P.R.; Waterman, M.S. (1978). "On some new sequences generalizing the Catalan and Motzkin numbers". Discrete Math. 26 (3): 261–272. doi:10.1016/0012-365x(79)90033-5.
- ↑ M.S. Waterman. Secondary structure of single - stranded nucleic acids. Adv. Math. I (suppl.), 1:167–212, 1978.
- 1 2 Smith, Temple F.; Waterman, Michael S. (1981). "Identification of common molecular subsequences". Journal of Molecular Biology. 147 (1): 195–197. doi:10.1016/0022-2836(81)90087-5. PMID 7265238.
- 1 2 Nussiniv; et al. (1978). "Algorithms for Loop Matchings". SIAM Journal on Applied Mathematics. 35 (1): 68–82. doi:10.1137/0135006. JSTOR 2101031.
- ↑ Howell, J.A.; Smith, T.F.; Waterman, M.S. (1980). "Computation of generating functions for biological molecules". SIAM J. Appl. Math. 39: 119133. doi:10.1137/0139010.
- ↑ Sankoff, David (October 1985). "Simultaneous Solution of the RNA Folding, Alignment and Protosequence Problems". SIAM Journal on Applied Mathematics. 45 (5): 810–825. doi:10.1137/0145048.
- ↑ Waterman, M.S.; Smith, T.F. (1986). "Rapid dynamic programming algorithms for RNA secondary structure". Adv. Appl. Math. 7 (4): 455–464. doi:10.1016/0196-8858(86)90025-4.
- ↑ McCaskill, John (1990). "The Equilibrium Partition Function and Base Pair Binding Probabilities for RNA Secondary Structure". Biopolymers. 29 (6–7): 1105–19. doi:10.1002/bip.360290621. hdl:11858/00-001M-0000-0013-0DE3-9. PMID 1695107. S2CID 12629688.
- ↑ Zuker, Michael; Stiegler, Patrick (1981). "Optimal Computer Folding of Large RNA Sequences Using Thermodynamics". Nucleic Acids Research. 9 (1): 133–148. doi:10.1093/nar/9.1.133. PMC 326673. PMID 6163133.
- ↑ Studnicka, Gary M.; Rahn, Georgia M.; Cummings, Ian W.; Salser, Winston A. (1978-09-01). "Computer method for predicting the secondary structure of single-stranded RNA". Nucleic Acids Research. 5 (9): 3365–3388. doi:10.1093/nar/5.9.3365. ISSN 0305-1048. PMC 342256. PMID 100768.
- ↑ Hofacker, I.L.; Fontana, W.; Stadler, P.F.; et al. (1994). "Fast folding and comparison of RNA secondary structures". Monatsh Chem. 125 (2): 167. doi:10.1007/BF00818163. S2CID 19344304.
- 1 2 Schuster, Peter; Fontana, Walter; Stadler, Peter F.; Hofacker, Ivo L. (1994-03-22). "From Sequences to Shapes and Back: A Case Study in RNA Secondary Structures". Proceedings of the Royal Society of London B: Biological Sciences. 255 (1344): 279–284. Bibcode:1994RSPSB.255..279S. doi:10.1098/rspb.1994.0040. ISSN 0962-8452. PMID 7517565. S2CID 12021473.
- ↑ "Neutral networks of RNA Secondary Structures" (PDF).
- ↑ Hofacker, Ivo L.; Schuster, Peter; Stadler, Peter F. (1998). "Combinatorics of RNA secondary structures". Discrete Applied Mathematics. 88 (1–3): 207–237. doi:10.1016/s0166-218x(98)00073-0.