The PDE2 (phosphodiesterase 2) enzyme is one of 21 different phosphodiesterases (PDE) found in mammals. These different PDEs can be subdivided to 11 families (PDE1 – PDE11). The different PDEs of the same family are functionally related despite the fact that their amino acid sequences show considerable divergence.[1] The PDEs have different substrate specificities. Some are cAMP selective hydrolases (PDE 4, -7 and -8), others are cGMP selective hydrolases (PDE 5, -6 and -9) and the rest can hydrolyse both cAMP and cGMP (PDE1, -2, -3, -10 and -11).[2]
There is only one gene family coding for the PDE2, which is the PDE2A. Three splice variants have been found, the PDE2A1, PDE2A2 and PDE2A3 (PDE2A2 has only been found in rats). PDE2A1 is cytosolic whereas -A2 and -A3 are membrane bound. It has been suggested that different localization of PDE2A2 and -A3 is due to a unique N-terminal sequence, which is absent in PDE2A1. Despite the PDE2A splice variants being different, there is no known differences in their kinetic behavior.[3]
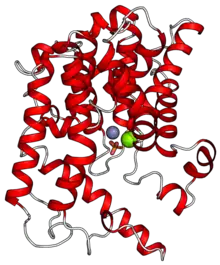
Crystal structure
The crystal structure of the active site of the PDE2 enzyme has been reported.[4] Even though amino acid sequences, for members of the PDE family show considerable difference (25-35% identity), the overall folding, functional and structural elements of the active sites are very similar. The active site is formed by residues that are highly conserved among all PDEs. The binding pocket contains metal ion (zinc and magnesium) binding sites. The two histidine and two aspartic acid residues, which bind zinc are conserved among all studied PDEs.[3] The structure of several other PDE iso-enzymes has been elucidated and among them few co-crystal structures, with inhibitors residing in the active site.[5] The co-crystal structures for PDE4B, PDE4D and PDE5A have revealed two common features of inhibitor binding to PDEs. One is a planar ring structure of the inhibitors, which align in the active site of the enzymes and the other is a conserved glutamine residue (the "glutamine switch" mentioned below), which is essential for nucleotide recognition and selectivity.[1][5][6]
Substrate selectivity
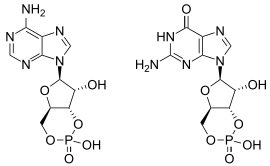
As mentioned above, PDE2 is able to hydrolyze both cAMP and cGMP, whereas some other members of the PDE family are selective for either of the two cyclic nucleotides. The variability in selectivity towards either cAMP or cGMP is thought to be determined by a so-called "glutamine switch". The "glutamine switch" is an invariant glutamine found in all PDEs, for which the crystal structure has been solved. In PDE2, this residue is the Gln859. It has potential to form hydrogen bonds with the exocyclic amino group of cAMP and the exocyclic carbonyl oxygen of cGMP. In PDEs, which can hydrolyze both cAMP and cGMP, this glutamine is able to rotate freely. In PDEs that are selective for either cAMP or cGMP, this glutamine is constrained by neighboring residues to a position favoring selectivity for either cyclic nucleotide.[1][3]
Regulation
When cGMP binds to the allosteric GAF-B domain of the PDE, it causes conformational change in the protein structure leading to higher enzyme activity. Increased hydrolysis of cAMP due to binding of cGMP to the GAF-B domain is well documented, however there are no known examples for the reverse.[3] It has been shown that the GAF-B domain has 30-100 fold lower affinity for cAMP than for cGMP.[7] This information combined with what is currently known about intracellular cAMP concentrations, renders it unlikely that activation of cGMP hydrolysis by cAMP can take place in vivo.[3]
Clinical value of PDE2
PDE2 is expressed in various tissues, for example: adrenal medulla, brain, heart, platelet, macrophages and endothelial cells. The enzyme is thought to be involved in regulating many different intracellular processes, such as:[3][8]
- aldosterone secretion from the adrenal gland
- intracellular concentrations of cAMP in cardiomyocytes and of cAMP and cGMP in platelets
- cGMP in neurons and effect on long-term memory
- barrier function of endothelial cells under inflammatory conditions
Several enzyme functions have been reported for the PDE2. It has been shown that PDE2 lowers cAMP through increased cGMP caused by atrial natriuretic peptide (ANP) resulting in decreased aldosterone secretion.[3] It has also been suggested that PDE2 might play an important role in the regulation of elevated intracellular concentrations of cAMP and cGMP in platelets. PDE3 is an important player in platelet aggregation. It has been reported that higher concentration of cGMP causes inhibition of PDE3, whereas it stimulates PDE2. Interplay between those two functions seems to mediate an opposing regulation of cAMP in platelets.[3] PDE2 regulates cardiac L-type Ca2+ current in cardiac myocytes, where activation of PDE2 by cGMP lowers cAMP and thereby affecting cardiac function.[3] However, it has been recently found that different cAMP pools, located within the cardiac myocyte, mediate different (moreover, sometimes opposing) effects. As different PDE types may affect different cAMP pools, the different PDEs may regulate different processes in the cell.[9] PDE2 is expressed in several regions of the brain and rat experiments have indicated that inhibition of PDE2 enhances functions such as memory.[3] PDE2 is up regulated when monocytes differentiate into macrophages, but the role of PDE2 in matured macrophages is yet to be characterized. Furthermore, PDE2 has been indicated to play a role in inflammatory responses as it has been detected in microvessels, but not in larger vessels. It has been speculated that tumor necrosis factor-alpha (TNFα) might regulate the function of PDE2 in endothelial cells and thereby affecting flow of fluid and cells through the endothelial barrier as in vitro experiments on endothelial cells show up regulation of both PDE2 mRNA and activity.[3]
Until now PDE2 inhibitors have mostly been used as research tools, but are presently being investigated for improving memory, for decreasing endothelial permeability under inflammatory conditions,[3] and for preventing/improving heart failure and cardiac hypertrophy.[10]
PDE2A inhibitors
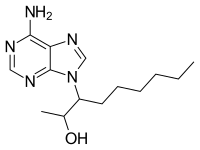

EHNA
The first specific inhibitor developed for PDE2 was EHNA (erythro-9-(2-hydroxy-3-nonyl)adenine). It has been demonstrated to specifically act on PDE2 by inhibiting cGMP-activation of PDE2 with an IC50 value of ~1μM and an at least 50-fold selectivity over other PDEs.[11] The core structure of EHNA resembles cAMP but differentiates in the fact that EHNA has a bulky hydrophobic carbon side chain replacing the phospho-ribose moiety in cAMP.[1]
Inhibitory effects of EHNA
In primary cultures of rat cortical neurons, the inhibition of PDE2A by EHNA potentiates NMDA (N-methyl-D-aspartate) receptor activated increase in cGMP, but has no effect on cAMP concentrations.[12] EHNA is also a very potent adenosine deaminase inhibitor with an IC50 ~2 nM.[13] This dual inhibition would lead to the accumulation of the two inhibitory metabolites, adenosine [13][14] and cGMP,[12][15] which may act in synergy to mediate diverse pharmacological responses including anti-viral, anti-tumour and anti-arrhythmic effects.[11] Although EHNA potently inhibits adenosine deaminase, it has been successfully used with the proper controls as a tool to probe PDE2 functions. EHNA has been used to study implication of PDE2 in calcium control in cardiac myocytes [16] and has shown to be effective to reverse hypoxic pulmonary vasoconstriction in perfused lung models.[17] EHNA has been therefore been used for two purposes:
- to serve as a lead structure for the rational design of more selective and potent PDE2 inhibitors, and
- to define some of PDE's biological targets.
However, the use of EHNA as a chemical tool in determining the pharmacological role of PDE2 is limited due to its low potency to inhibit PDE2 and high potency in inhibition of adenosine deaminase.[18] Theoretically, this problem can be resolved if the effect of adenosine accumulated by EHNA, a result of adenosine deaminase inhibition, can be accounted and corrected for. This way, a positive inotropic effect elicited by EHNA, as a result of PDE2 inhibition, was observed.[19][20]
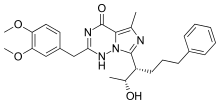
BAY 60-7550, oxindole and PDP
BAY 60-7550 is an analog of EHNA, which is more than 100-fold more potent and is highly selective for PDE2A.[13] Other newly discovered selective PDE2 inhibitors are PDP (9-(6-phenyl-2-oxohex-3-yl)-2-(3,4-dimethoxybenzyl)-purin-6-one) [21] and oxindole.[18]
| Inhibitor | IC50 | Reference |
| EHNA | 1 μM | [11] |
| Oxindole | 40 nM | [16] |
| Bay 60-7550 | 4,7 nM | [13] |
| PDP | 0,6 nM | [21] |
The table above shows the potency of PDE2 inhibitors including EHNA. There is a large increase in potency between EHNA, Bay 60-7550 and PDP. The large dimethoxybenzyl group in position 2 of the purine moiety of Bay-60 7550 and PDP might be contributing to the added potency.
Structure and bonding of the inhibitors
Comparison of these inhibitors with the natural substrates of the enzyme, cAMP and cGMP reveal some common characteristics of the molecules. The main characteristic of all the molecules is the flat moiety comprising at least two fused ring structures, a six atom ring and a five atom ring. This ring system in cGMP and cAMP is a purine ring system, and the same is true for EHNA and PDP. Bay 60-7550 and oxindole lack the purine core but do possess a related ring system. Hydrogen bond acceptors, mostly nitrogen but also oxygen, reside in the ring system of the inhibitors. These atoms might interact with hydrogen bond donators, which are part of amino acids in the active site of the enzyme and thereby contribute to the inhibition of the enzyme from hydrolyzing cAMP and cGMP similar to how the natural substrates bind to the active site.[1]
Structural similarity of the inhibitors
The structures of Bay 60-7550 and PDP are very similar. The difference between these molecules is the exocyclic methyl group on Bay 60-7550, which replaces the nitrogen atom in PDP decreasing the possibility to form hydrogen bonds with the enzyme in an important site for substrate and inhibitor binding. The oxindole structure differs from the other inhibitors since it is more divergent from the purine ring system and has less hydrogen binding possibilities. The molecule also lacks the large side group, analogous to the dimethoxybenzyl group of Bay 60-7550 and PDP. It is difficult to predict possible interactions to the enzyme without a co-crystal structure of the phenomenon.
Possible structure-activity relationship for PDE2 inhibitors
There is a lack of co-crystal structures of inhibitors bound in the active site of PDE2. However, a computerized docking model of the inhibitor EHNA and the substrates cAMP and cGMP bound in the catalytic site have been made.[1] The docking model of EHNA showed that the mutations of the amino acids Asp811 to Ala (Asp811Ala) and Ile826 to Val (Ile826Val) at the active site, where the only amino acid substitutions that significantly affected the inhibition by EHNA. The Asp811 mutation to alanine increased IC50 value for EHNA 6-fold and the Ile826 mutation to valine leads to a 7-fold increased IC50 value for EHNA compared to wild type PDE2A.[1]
Upper binding pocket: Gln859 and Asp811
EHNA is in close proximity to Gln859 at the active site, which could donate two hydrogen bonds to N1 and N6 of the nitrogen atoms in the adenine ring of EHNA. On the other site of the binding pocket the Asp811 could donate another hydrogen bond to N7 in the adenine ring in order to stabilize the bond inhibitor. This hypothesis is supported by the fact that the Asp811Ala mutant has decreased activity toward cAMP, whereas activity toward cGMP is unchanged.[1]
The lower binding pocket: Ile 826
The residues in the lower binding pocket may lie too far away for interaction with the inhibitor and therefore might be irrelevant for EHNA selectivity. However the residues may play an indirect role of EHNA selectivity. Ile826 is positioned below the purine ring of EHNA and thereby limits the space for EHNA. Substitution with smaller valine (Ile826Val mutation) could increase the space for EHNA and cause the loss of hydrogen binding with residues in the upper binding pocket, while improving hydrogen binding within the lower binding pocket. This shift of interactions could destabilize binding of the adenine ring of EHNA, which could be the reason for higher IC50 value.[1]
No models are available for the other inhibitors than EHNA, which align in the active site. Therefore, it is more difficult to interpret the molecular binding. When looking at the inhibitors and their overall similarity, it is likely that they bind with a similar mechanism to the active site and that the different side groups determine potency of the inhibitor. The determinants of inhibitor specificity within the PDE2 active site are not very well known and with better understanding of these determinants it would facilitate the development of inhibitors with increased potency.[1]
References
- 1 2 3 4 5 6 7 8 9 10 Iffland A, Kohls D, Low S, et al. (June 2005). "Structural determinants for inhibitor specificity and selectivity in PDE2A using the wheat germ in vitro translation system". Biochemistry. 44 (23): 8312–25. doi:10.1021/bi047313h. PMID 15938621.
- ↑ Martinez SE, Wu AY, Glavas NA, et al. (October 2002). "The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding". Proc. Natl. Acad. Sci. U.S.A. 99 (20): 13260–5. doi:10.1073/pnas.192374899. PMC 130621. PMID 12271124.
- 1 2 3 4 5 6 7 8 9 10 11 12 Bender AT, Beavo JA (September 2006). "Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use". Pharmacol. Rev. 58 (3): 488–520. doi:10.1124/pr.58.3.5. PMID 16968949. S2CID 7397281.
- ↑ PDB: 1Z1L
- 1 2 Card GL, England BP, Suzuki Y, et al. (December 2004). "Structural basis for the activity of drugs that inhibit phosphodiesterases". Structure. 12 (12): 2233–47. doi:10.1016/j.str.2004.10.004. PMID 15576036.
- ↑ Zhang KY, Card GL, Suzuki Y, et al. (July 2004). "A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases". Mol. Cell. 15 (2): 279–86. doi:10.1016/j.molcel.2004.07.005. PMID 15260978.
- ↑ Wu AY, Tang XB, Martinez SE, Ikeda K, Beavo JA (September 2004). "Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A". J. Biol. Chem. 279 (36): 37928–38. doi:10.1074/jbc.M404287200. PMID 15210692.
- ↑ Kass DA (2013). "Et Tu PDE2?". J Am Coll Cardiol. 62 (17): 1607–1609. doi:10.1016/j.jacc.2013.06.003. PMID 23810892.
- ↑ Stangherlin A; Gesellchen F; Zoccarato A; Terrin A; Fields LA; Berrera M; Surdo NC; Craig MA; Smith G; Hamilton G; Zaccolo M (2011). "cGMP signals modulate camp levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes". Circ Res. 108 (8): 929–939. doi:10.1161/CIRCRESAHA.110.230698. PMC 3083836. PMID 21330599.
- ↑ Zoccarato A; Surdo NC; Aronsen JM; Fields LA; Mancuso L; Dodoni G; Stangherlin A; Livie C; Jiang H; Sin YY; Gesellchen F; Terrin A; Baillie GS; Nicklin SA; Graham D; Szabo-Fresnais N; Krall J; Vandeput F; Movsesian M; Furlan L; Corsetti V; Hamilton GM; Lefkimmiatis K; Sjaastad I; Zaccolo M (2015). "Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2" (PDF). Circ Res. 117 (8): 707–719. doi:10.1161/CIRCRESAHA.114.305892. PMID 26243800.
- 1 2 3 Podzuweit T, Nennstiel P, Müller A (September 1995). "Isozyme selective inhibition of cGMP-stimulated cyclic nucleotide phosphodiesterases by erythro-9-(2-hydroxy-3-nonyl) adenine". Cell. Signal. 7 (7): 733–8. doi:10.1016/0898-6568(95)00042-N. PMID 8519602.
- 1 2 Suvarna NU, O'Donnell JM (July 2002). "Hydrolysis of N-methyl-D-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus". J. Pharmacol. Exp. Ther. 302 (1): 249–56. doi:10.1124/jpet.302.1.249. PMID 12065724.
- 1 2 3 4 Boess FG, Hendrix M, van der Staay FJ, et al. (December 2004). "Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance". Neuropharmacology. 47 (7): 1081–92. doi:10.1016/j.neuropharm.2004.07.040. PMID 15555642. S2CID 25756665.
- ↑ van Calker D, Müller M, Hamprecht B (1978). "Adenosine inhibits the accumulation of cyclic AMP in cultured brain cells". Nature. 276 (5690): 839–41. doi:10.1038/276839a0. PMID 214714. S2CID 4272425.
- ↑ Goldberg ND, Haddox MK, Nicol SE, et al. (1975). "Biologic regulation through opposing influences of cyclic GMP and cyclic AMP: the Yin Yang hypothesis". Adv Cyclic Nucleotide Res. 5: 307–30. PMID 165672.
- 1 2 Rivet-Bastide M, Vandecasteele G, Hatem S, et al. (June 1997). "cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes" (PDF). J. Clin. Invest. 99 (11): 2710–8. doi:10.1172/JCI119460. PMC 508117. PMID 9169501.
- ↑ Haynes J, Killilea DW, Peterson PD, Thompson WJ (February 1996). "Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic-3',5'-guanosine monophosphate-stimulated phosphodiesterase to reverse hypoxic pulmonary vasoconstriction in the perfused rat lung". J. Pharmacol. Exp. Ther. 276 (2): 752–7. PMID 8632346.
- 1 2 Chambers RJ, Abrams K, Garceau NY, et al. (January 2006). "A new chemical tool for exploring the physiological function of the PDE2 isozyme". Bioorg. Med. Chem. Lett. 16 (2): 307–10. doi:10.1016/j.bmcl.2005.10.005. PMID 16275071.
- ↑ Gesztelyi R; Zsuga J; Hajdu P; Szabo JZ; Cseppento A; Szentmiklosi AJ (2003). "Positive inotropic effect of the inhibition of cyclic GMP-stimulated 3',5'-cyclic nucleotide phosphodiesterase (PDE2) on guinea pig left atria in eu- and hyperthyroidism" (PDF). Gen Physiol Biophys. 22 (4): 501–513. PMID 15113122.
- ↑ Kemeny-Beke A; Jakab A; Zsuga J; Vecsernyes M; Karsai D; Pasztor F; Grenczer M; Szentmiklosi AJ; Berta A; Gesztelyi R (2007). "Adenosine deaminase inhibition enhances the inotropic response mediated by A1 adenosine receptor in hyperthyroid guinea pig atrium". Pharmacol Res. 56 (2): 124–131. doi:10.1016/j.phrs.2007.04.017. PMID 17574432.
- 1 2 Seybold J, Thomas D, Witzenrath M, et al. (May 2005). "Tumor necrosis factor-alpha-dependent expression of phosphodiesterase 2: role in endothelial hyperpermeability". Blood. 105 (9): 3569–76. doi:10.1182/blood-2004-07-2729. PMID 15650061.