Stimulated Raman spectroscopy, also referred to as stimulated Raman scattering (SRS) is a form of spectroscopy employed in physics, chemistry, biology, and other fields. The basic mechanism resembles that of spontaneous Raman spectroscopy: a pump photon, of the angular frequency , which is scattered by a molecule has some small probability of inducing some vibrational (or rotational) transition, as opposed to inducing a simple Rayleigh transition. This makes the molecule emit a photon at a shifted frequency. However, SRS, as opposed to spontaneous Raman spectroscopy, is a third-order non-linear phenomenon involving a second photon—the Stokes photon of angular frequency —which stimulates a specific transition. When the difference in frequency between both photons () resembles that of a specific vibrational (or rotational) transition () the occurrence of this transition is resonantly enhanced. In SRS, the signal is equivalent to changes in the intensity of the pump and Stokes beams. The signals are typically rather low, of the order of a part in 10^5, thus calling for modulation-transfer techniques: one beam is modulated in amplitude and the signal is detected on the other beam via a lock-in amplifier. Employing a pump laser beam of a constant frequency and a Stokes laser beam of a scanned frequency (or vice versa) allows for the unraveling of the spectral fingerprint of the molecule. This spectral fingerprint differs from those obtained by other spectroscopy methods such as Rayleigh scattering as the Raman transitions confer to different exclusion rules than those that apply for Rayleigh transitions.




History
The phenomenon of SRS was discovered by an accident by Woodbury and Ng in 1962.[1] In their experiment, they introduced a Kerr cell containing nitrobenzene into a ruby laser cavity to study Q-switching processes. This resulted with a strong emission at a wavelength in the IR region that could not be associated with the characteristic wavelengths of the ruby gain medium. At first, this was explained as luminescence. Only at a later stage was this interpreted correctly as the first experimental observation of SRS. A year later, Garmier et al.[1] introduced a two-wave mixing framework for the description of SRS. These pioneering works opened a new avenue of research and were followed by many theoretical and experimental works in the field of SRS.
Principle
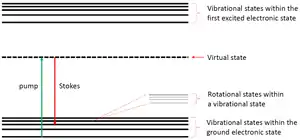
Qualitative description
The principle of SRS can be intuitively understood by adopting the quantum mechanical description of the molecule's energy levels. Initially, the molecule lies in the ground state, that is, its lowest electronic energy level. Then, it simultaneously absorbs both pump and Stokes photons, which causes a vibrational (or rotational) transition with some probability. The transition can be thought of as a two-step transition where in the first step the molecule is excited by the pump photon to a virtual state and in the second it is relaxed into a vibrational (or rotational) state other than the ground state. The virtual state, which is actually a superposition of probability tails of real states, cannot be occupied by the molecule. However, for a simultaneous absorption of two photons, it might provide a coupling route between the initial and final states. When the energy difference between both pump and Stokes photons matches the energy difference between some vibrational (or rotational) state and the ground state, the probability for a transition due to this stimulated process is enhanced by orders of magnitude.
Quantitative description
Each photon that undergoes SRS is shifted in color from pump to Stokes color. Thus, the SRS signal is proportional to the decrease or increase in the pump or Stokes beams intensities, respectively. These changes in the beams intensities are described by the following rate equations


where, and are the pump and Stokes beams intensities, respectively, and are the pump and Stokes angular frequencies, respectively, is the coordinate along which the beams propagate, is the Raman gain coefficient, and is the loss coefficient. The loss coefficient is an effective coefficient that might account for loses due to a variety of processes such as Rayleigh scattering, absorption, etc. The first rate equation describes the change in Stokes beam intensity along the SRS interaction length. The first term on the right-hand side is equivalent to the amount of intensity gained by the Stokes beam due to SRS. As SRS involves both beams, this term is dependent both on and . The second term is equivalent to the amount of intensity lost and is thus dependent only on . The second rate equation describes the change in pump beam intensity, its form is quite similar to the former. The first term on the right hand side of the second equation equals its counterpart from the first equation up to a multiplicative factor of . This factor reflects the fact that each photon (as opposed to intensity units) lost from the pump beam due to SRS is gained by the Stokes beam.






In most cases, the experimental conditions support two simplifying assumptions: (1) photon loss along the Raman interaction length, , is negligible. Mathematically this corresponds to


and (2) the change in beam intensity is linear; mathematically this corresponds to
- .

Accordingly, the SRS signal, that is, the intensity changes in pump and Stokes beams is approximated by


where and are the initial pump and Stokes beams intensities, respectively. As for the Raman interaction length, in many cases this length can be evaluated in a similar fashion to the evaluation of the Rayleigh length as


- .

Here, and are the averaged refractive index and beam waist, respectively, and and are the pump and Stokes wavelengths, respectively.




Every molecule has some characteristic Raman shifts, each associated with a specific vibrational (or rotational) transition of the molecule. The relation between a Raman shift, , and the pump and Stokes photon wavelengths is given by

![{\displaystyle \Delta \omega [\mathrm {cm} ^{-1}]=\left({\frac {1}{\lambda _{p}[\mathrm {nm} ]}}-{\frac {1}{\lambda _{S}[\mathrm {nm} ]}}\right)\times {\frac {[10^{7}\mathrm {nm} ]}{[\mathrm {cm} ]}}}](../I/0bb109a2386e2a6519cd5ad8084b30b16a5dd376.svg)
When the difference in wavelengths between both lasers is close to some Raman transition, the Raman gain coefficient receives values on the order of resulting with an efficient SRS. As this difference starts to differ from a specific Raman transition the value of the Raman gain coefficient drops down and the process becomes increasingly less efficient and less detectable.
![{\displaystyle 10^{-30}[\mathrm {cm} ^{2}/\mathrm {molecule} \cdot \mathrm {sr} ]}](../I/0538e0dce3f76d829efc47357e20295fd662fb45.svg)
An SRS experimental setup includes two laser beams (usually co-linear) of the same polarization, one is employed as pump and the other as Stokes. Usually, at least one of the lasers is pulsed. This modulation in the laser intensity helps to detect the signal, furthermore, it helps increase the signal's amplitude, which also helps detection. When designing the experimental setup, one has great liberty when choosing the pump and Stokes lasers as the Raman condition (shown in the equation above) applies only on the difference in wavelengths.
Comparison with other Raman spectroscopy variants
Since SRS is a resonantly enhanced process, its signal is several orders of magnitude higher than that of a spontaneous Raman scattering, making it a much more efficient spectroscopic tool. Furthermore, the signal intensity of SRS is also several orders of magnitude higher than another highly common sort of spectroscopy – coherent anti-Stokes Raman spectroscopy. SRS involves only two photons, as opposed to the latter, which involves three photons. Thus, the occurrence of SRS is more probable and results with a higher signal. There are two additional prominent variants of spontaneous Raman spectroscopy – surface-enhanced Raman spectroscopy and resonance Raman spectroscopy. The former is designated for Raman spectroscopy of molecules adsorbed on rough surfaces such as metal surfaces or nanostructures where it magnifies the Raman signal by many orders of magnitude.[2] The latter corresponds to a spontaneous Raman scattering process performed by a laser with a frequency close to the electronic transition of the subject in study. This may amplify the signal. However, it requires the use of highly powerful UV or X-ray lasers that might cause photodegradation and might also induce fluorescence.
Applications
SRS is employed in various applications from a wide variety of fields. All applications utilize the ability of SRS to detect a vibrational (or rotational) spectral signature of the subject in study. Here are some examples:
Study of molecular conformational structures
Works in this field were done both in the Cina[3] and Bar[4][5] groups. Each conformer is associated with a slightly different SRS spectral signature. Detection of these different landscapes is an indication of the different conformational structures of the same molecule.
Material composition analysis
Here, the SRS signal dependence on the concentration of the material is utilized. Measuring different SRS signals, associated with the different materials in the composition, allow for the determination of the stoichiometric relations of the composition.
Microscopy
Stimulated Raman scattering (SRS) microscopy allows non-invasive label-free imaging in living tissue. In this method, pioneered by the Xie group,[6] a construction of an image is obtained by performing SRS measurements over some grid, where each measurement adds a pixel to the image.
Ultrafast microscopy
Employing femtosecond laser pulses, as was done in the Katz, Silberberg,[7] and Xie[8] groups, allows for an instant generation of a very large portion of the spectral signature by a single laser pulse. The broad signal is a result of the width of the laser band as dictated by the uncertainty principle, which determines an inverse proportion between the uncertainty in time and uncertainty in frequency. This method is far faster than traditional microscopy methods as it circumvents the need in long and time-consuming frequencies scanning.
See also
References
- 1 2 Prince, R.C.; Frontiera, R.R.; Potma, E.O. (2017). "Stimulated Raman Scattering: From Bulk to Nano". Chem. Rev. 117 (7): 5070−5094. doi:10.1021/acs.chemrev.6b00545. PMC 5471143. PMID 27966347.
- ↑ Xu, X.; Li, H.; Hansan, D.; Ruoff, R.S.; Wang, A.X.; Fan, D.L. (2013). "Near-Field Enhanced Plasmonic-Magnetic Bifunctional Nanotubes for Single Cell Bioanalysis". Adv. Funct. Mater. 23 (35): 4332–4338. doi:10.1002/adfm.201203822.
- ↑ Cina, J.A.; Kovac, P.A. (2013). "How Fissors Works: Observing Vibrationally Adiabatic Conformational Change through Femtosecond Stimulated Raman Spectroscopy". J. Phys. Chem. A. 117 (29): 6084−6095. Bibcode:2013JPCA..117.6084C. doi:10.1021/jp312878t. PMID 23590752.
- ↑ Mayorkas, N.; Bernat, A.; Izbitski, S.; Bar, I. (2012). "Simultaneous Ionization-Detected Stimulated Raman and Visible–Visible–Ultraviolet Hole-Burning Spectra of Two Tryptamine Conformers". J. Phys. Chem. Lett. 3 (5): 603–607. doi:10.1021/jz300026a. PMID 26286155.
- ↑ Mayorkas, N.; Bernat, A.; Izbitski, S.; Bar, I. (2013). "Vibrational and vibronic spectra of tryptamine conformers". J. Chem. Phys. 138 (12): 124312. Bibcode:2013JChPh.138l4312M. doi:10.1063/1.4798218. PMID 23556728.
- ↑ Freudiger, C.W.; Min, W.; Saar, B.G.; Lu, S.; Holtom, G.R.; He, C.; Tsai, J.C.; Kang, J.X.; Xie, X.S. (2008). "Label-Free Biomedical Imaging with High Sensitivity by Stimulated Raman Scattering Microscopy". Science. 322 (5909): 1857–1861. Bibcode:2008Sci...322.1857F. doi:10.1126/science.1165758. PMC 3576036. PMID 19095943.
- ↑ Frostig, H.; Katz, O.; Natan, A.; Silberberg, Y. (2011). "Single-pulse stimulated Raman scattering spectroscopy". Optics Letters. 36 (7): 1248–1250. arXiv:1011.6576. Bibcode:2011OptL...36.1248F. doi:10.1364/OL.36.001248. PMID 21479047.
- ↑ Fu, D.; Holtom, G.; Freudiger, C.; Zhang, X.; Xie, X.S. (2013). "Hyperspectral Imaging with Stimulated Raman Scattering by Chirped Femtosecond Lasers". J. Phys. Chem. B. 117 (16): 4634–4640. doi:10.1021/jp308938t. PMC 3637845. PMID 23256635.
Further reading
- Kazzaz, A.; Ruschin, S.; Shoshan, I.; Ravnitsky, G. (1994). "Stimulated Raman Scattering in Methane-Experimental Optimization and Numerical Model". IEEE Journal of Quantum Electronics. 30 (12): 3017−3024. Bibcode:1994IJQE...30.3017K. doi:10.1109/3.362703.
- Tipping, W.J.; Lee, M.; Serrels, A.; Brunton, V.G.; Hulme, A.N. (2016). "Stimulated Raman scattering microscopy: an emerging tool for drug discovery". Chem. Soc. Rev. 45 (8): 2075–2089. doi:10.1039/c5cs00693g. PMC 4839273. PMID 26839248.