A total internal reflection fluorescence microscope (TIRFM) is a type of microscope with which a thin region of a specimen, usually less than 200 nanometers can be observed.
TIRFM is an imaging modality which uses the excitation of fluorescent cells in a thin optical specimen section that is supported on a glass slide. The technique is based on the principle that when excitation light is totally internally reflected in a transparent solid coverglass at its interface with a liquid medium, an electromagnetic field, also known as an evanescent wave, is generated at the solid-liquid interface with the same frequency as the excitation light.[1] The intensity of the evanescent wave exponentially decays with distance from the surface of the solid so that only fluorescent molecules within a few hundred nanometers of the solid are efficiently excited. Two-dimensional images of the fluorescence can then be obtained, although there are also mechanisms in which three-dimensional information on the location of vesicles or structures in cells can be obtained.[2]
History
Widefield fluorescence was introduced in 1910 which was an optical technique that illuminates the entire sample.[3] Confocal microscopy was then introduced in 1960 which decreased the background and exposure time of the sample by directing light to a pinpoint and illuminating cones of light into the sample. In the 1980s, the introduction of TIRFM further decreased background and exposure time by only illuminating the thin section of the sample being examined.[3]
Background
There are two common methods for producing the evanescent wave for TIRFM.[1] The first is the prism method which uses a prism to direct the laser toward the interface between the coverglass and the media/cells at an incident angle sufficient to cause total internal reflection. This configuration has been applied to cellular microscopy for over 30 years but has never become a mainstream tool due to several limitations. Although there are many variations of the prism configuration, most restrict access to the specimen which makes it difficult to perform manipulations, inject media into the specimen space, or carry out physiological measurements.[2] Another disadvantage is that in most configurations based on the inverted microscope designs, the illumination is introduced on the specimen side opposite of the objective optics which requires imaging of the evanescent field region through the bulk of the specimen. There is great complexity and precision required in imaging this system which meant that the prism method was not used by many biologists but rather limited to use by physicists.
The other method is known as the objective lens method which has increased the use of TIRFM in cellular microscopy and increased furthermore since a commercial solution became available.[2] In this mechanism, one can easily switch between standard widefield fluorescence and TIRF by changing the off-axis position of the beam focus at the objective's back focal plane. There are several developed ways to change the positions of the beam such as using an actuator that can change the position in relation to the fluorescence illuminator that is attached to the microscope.
Application
In cell and molecular biology, a large number of molecular events in cellular surfaces such as cell adhesion, binding of cells by hormones, secretion of neurotransmitters, and membrane dynamics have been studied with conventional fluorescence microscopes. However, fluorophores that are bound to the specimen surface and those in the surrounding medium exist in an equilibrium state. When these molecules are excited and detected with a conventional fluorescence microscope, the resulting fluorescence from those fluorophores bound to the surface is often overwhelmed by the background fluorescence due to the much larger population of non-bound molecules. TIRFM allows for selective excitation of the surface-bound fluorophores, while non-bound molecules are not excited and do not fluoresce. Due to the fact of sub-micron surface selectivity, TIRFM has become a method of choice for single molecule detection.
There are many applications of TIRFM in cellular microscopy. Some of these applications include:
- Measuring the kinetics of receptor endocytosis in response to ligand binding and receptor movement
- Observing exocytic events through the loading of vesicles undergoing exocytosis with fluorescent dyes
- Qualitatively and quantitatively describing the roles different proteins play in exocytosis/endocytosis
- Observing the size, movement, and distance apart of the regions of contact between a cell and a solid substrate
With the ability to resolve individual vesicles optically and follow the dynamics of their interactions directly, TIRFM provides the capability to study the vast number of proteins involved in neurobiological processes in a manner that was not possible before.[2]
Benefits
TIRFM provides several benefits over standard widefield and confocal fluorescence microscopy such as:
- The background is substantially decreased so structures can be seen clearly
- There is virtually no out-of-focus fluorescence collected which decrease blurring effects
- Cells are exposed to a significantly smaller amount of light which limits phototoxicity to cells
Overview
The idea of using total internal reflection to illuminate cells contacting the surface of glass was first described by E.J. Ambrose in 1956.[4] This idea was then extended by Daniel Axelrod[5] at the University of Michigan, Ann Arbor in the early 1980s as TIRFM. A TIRFM uses an evanescent wave to selectively illuminate and excite fluorophores in a restricted region of the specimen immediately adjacent to the glass-water interface. The evanescent electromagnetic field decays exponentially from the interface, and thus penetrates to a depth of only approximately 100 nm into the sample medium. Thus the TIRFM enables a selective visualization of surface regions such as the basal plasma membrane (which are about 7.5 nm thick) of cells. Note, however, that the region visualized is at least a few hundred nanometers wide, so the cytoplasmic zone immediately beneath the plasma membrane is necessarily visualized in addition to the plasma membrane during TIRF microscopy. The selective visualization of the plasma membrane renders the features and events on the plasma membrane in living cells with high axial resolution.
TIRF can also be used to observe the fluorescence of a single molecule,[6][7] making it an important tool of biophysics and quantitative biology. TIRF microscopy has also been applied in the single molecule detection of DNA biomarkers and SNP discrimination.[8]
Cis-geometry (through-objective TIRFM) and trans-geometry (prism- and lightguide based TIRFM) have been shown to provide different quality of the effect of total internal reflection. In the case of trans-geometry, the excitation lightpath and the emission channel are separated, while in the case of objective-type TIRFM they share the objective and other optical elements of the microscope. Prism-based geometry was shown to generate clean evanescent wave, which exponential decay is close to theoretically predicted function.[9] In the case of objective-based TIRFM, however, the evanescent wave is contaminated with intense stray light. The intensity of stray light was shown to amount 10–15% of the evanescent wave, which makes it difficult to interpret data obtained by objective-type TIRFM[10][11]
Mechanism
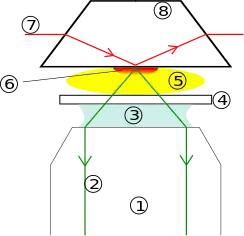
- Objective
- Emission beam (signal)
- Immersion oil
- Cover slip
- Specimen
- Evanescent wave range
- Excitation beam
- Quartz prism
The basic components of the TIRFM device include:
- Excitation beam light source
- Cover slip and immersion oil
- Objective lens
- Sample specimen
- Detector
Objective-based vs prism-based
Key differences between objective-based (cis) and prism-based (trans) TIRFM are that prism based TIRFM requires usage of a prism/solution interface to generate the evanescent field, while objective-based TIRFM does not require a prism and utilizes a cover slip/solution interface to generate the evanescent field. Typically objective-based TIRFM are more popularly used, however have lowered imaging quality due to stray light noise within the evanescent wave.
Prism-based
- Less extraneous scattering
- Much cheaper (hundreds instead of thousands of dollars)
- Best for low-mid range magnification and water immersion objectives
- Easiest with free collimated laser sources
- Larger range of incidence angles
- Desirable to achieve smallest evanescent field depth

- Specimen
- Evanescent wave range
- Cover slip
- Immersion oil
- Objective
- Emission beam (signal)
- Excitation beam
Objective-based
- High magnification and aperture
- Stable, easy to set up and align
- Works with free collimated laser, optical fiber, or conventional arc sources
Methodology
Fundamental physics
TIRFM is predicated on the optical phenomena of total internal reflection, in which waves arriving at a medium interface do not transmit into medium 2 but are completely reflected back into medium 1. Total internal reflection requires medium 2 to have a lower refractive index than medium 1, and for the waves must be incident at sufficiently oblique angles on the interface. An observed phenomena accompanying total internal reflection is the evanescent wave, which spatially extends away perpendicularly from the interface into medium 2, and decays exponentially, as a factor of wavelength, refractive index, and incident angle. It is the evanescent wave which is used to achieve increased excitation of the fluorophores close to the surface of the sample, and diminished excitation of superfluous fluorophores within solution.
For practical purposes, in objective based TIRF, medium 1 is typically a high refractive index glass coverslip, and medium 2 is the sample in solution with a lower refractive index. There may be immersion oil between the lens and the glass coverslip to prevent significant refraction through air.
Evanescent wave
The critical angle for excitatory light incidence can be derived from Snell's law:

For the refractive index of sample, the refractive index of the cover slip.


Thus, as the angle of incidence reaches , we begin observing effects of total internal reflection and evanescent wave, and as it surpasses these effects are more prevalent.

The intensity of the evanescent wave is given by:

With penetration depth given by:


Typically, ≤~100 nanometers, which is typically much smaller than the wavelength of light, and much thinner than a slice from confocal microscopes.[1][12]
For TIRFM imaging the wavelength of the excitation beam within the sample can be selected for by filtering. Additionally, the range of incident angles is determined by the numerical aperture (NA) of the objective, and requires that NA > . This parameter can be adjusted by changing the angle the excitation beam enters the objective lens. Finally, the reflective indices () of the solution and cover slip can be experimentally found or reported by manufacturers.



Excitation beam
For complex fluoroscope microscopy techniques, lasers are the preferred light source as they are highly uniform, intense, and near-monochromatic. However, it is noted that ARC LAMP light sources and other types of sources may also work. Typically the wavelength of excitation beam is designated by the requirements of the fluorophores within the sample, with most common excitation wavelengths being in the 400–700 nm range for biological samples.
In practice, a lightbox will generate a high intensity multichromatic laser, which will then be filtered to allow the desired wavelengths through to excite the sample. For objective-based TIRFM, the excitation beam and fluoresced emission beam will be captured via the same objective lens. Thus, to split the beams, a dichromatic mirror is used to reflect the incoming excitation beam towards the objective lens, and allow the emission beam to pass through into the detector. Additional filtering may be required to further separate emission and excitation wavelengths.
Emission beam
When excited with specific wavelengths of light, fluorophore dyes will reemit light at longer wavelengths (which contain less energy). In the context of TIRFM, only fluorophores close to the interface will be readily excited by the evanescent field, while those past ~100 nm will be highly attenuated. Light emitted by the fluorophores will be undirected, and thus will pass through the objective lens at varying locations with varying intensities. This signal will then pass through the dichromatic mirror and onward to the detector.
Cover slip and immersion oil
Glass cover slips typically have a reflective index around , while the immersion oil refractive index is a comparable . The medium of air, which has a refractive index of , would cause refraction of the excitation beam between the objective and the coverslip, thus the oil is used to buffer the region and prevent superfluous interface interactions before the beam reaches the interface between coverslip and sample.



Objective lens
The objective lens numerical aperture (NA) specifies the range of range of angles over which the system can accept or emit light.
To achieve the greatest incident angles, it is desirable to pass light at an off-axis angle through the peripheries of the lens.
Back focal plane (BPF)
The back focal plane (also called "aperture plane") is the plane through which the excitatory beam is focused before passing through the objective. Adjusting the distance between the objective and BPF can yield different imaging magnification, as the incident angle will become less or more steep. The beam must be passed through the BPF off-axis in order to pass through the objective at its ends, allowing for the angle to be sufficiently greater than the critical angle. The beam must also be focused at the BPF because this ensures that the light passing through the objective is collimated, interacting with the cover slip at the same angle and thus all totally internally reflecting.[12]
Sample
The sample should be adsorbed to the surface of the glass cover slide and stained with appropriate fluorophores to resolve the features desired within the sample. This is in protocol with any other fluorescent microscopy technique.
Dichroic (dichromatic) filter
The dichroic filter is an edge filter used at an oblique angle of incidence (typically 45°) to efficiently reflect light in the excitation band and to transmit light in the emission band. The 45° angle of the filter separates the path of the excitation and emission beam. The filter is composed of a complex system of multiple layers of metals, metal salts and dielectrics which have been vacuum-deposited onto thin glass.[13] This coating is designed to have high reflectivity for shorter wavelengths and high transmission for longer wavelengths.[14] While the filter transmits the selected excitation light (shorter wavelength) through the objective and onto the plane of the specimen, it also passes emission fluorescence light (longer wavelength) to the barrier filter and reflecting any scattered excitation light back in the direction of the laser source.[15] This maximizes the amount of exciting radiation passing through the filter and emitted fluorescence beam that is detected by the detector.[16]
Barrier filter
The barrier filter mainly blocks off undesired wavelengths, especially shorter excitation light wavelengths. It is typically a bandpass filter that passes only the wavelengths emitted by the fluorophore and blocks all undesired light outside this band. More modern microscopes enable the barrier filter to be changed according to the wavelength of the fluorophore's specific emission.[13]
Image detection and resolution
The image is detected by a charged-coupled device (CCD) digital camera. CCD cameras have photon detectors, which are thin silicon wafers, assembled into 2D arrays of light-sensitive regions. The detector arrays capture and store image information in the form of localized electrical charge that varies with incident light intensity.[2] As shown in the schematic the photons are transform to electrons by the detectors and the electrons are converted to readable electrical signal in the circuit board.[17] The electrical signal is then convoluted with a point spread function (PSF) to sample the original signal. As such, image resolution is highly dependent on the number of detectors and the point spread function will determine the image resolution.[18]
Image artifact and noise
Most fluorescence imaging techniques exhibit background noise due to illuminating and reconstructing large slices (in the z-direction) of the samples. Since TIRFM uses an evanescent wave to fluoresce a thin slice of the sample, there is inherently less background noise and artifacts. However, there are still other noises and artifacts such as poisson noise, optical aberrations, photobleaching, and other fluorescence molecules.
Poissonian noise are fundamental uncertainties with the measurement of light. This will cause uncertainties during the detection of fluorescence photons.[18] If N photons are measured in a particular measurement, there is a 63% probability that the true average value is in the range between N +√N and N −√N.[19] This noise may cause misrepresentation of the object at incorrect pixel locations.
Optical aberrations can arise from diffraction of fluorescence light or microscope and objective misalignment. Diffraction of light on the sample slide can spread the fluorescence signal and result in blurring in the convoluted images. Similarly, if there is a misalignment between the objective lens, filter, and detector, the excitation or emission beam may not be in focus and can cause blurring in the images.[14]
Photobleaching can occur when the covalent or noncovalent bonds in the fluorophores are destructed by the excitation light and can no longer fluoresce.[20] The fluorescing substances will always degrade to some extent by the energy of the exciting radiation and will cause the fluorescence to fade and result in a dark blurry image.[21] Photobleaching is inevitable but can be minimized by avoiding unwanted light exposure and using immersion oils to minimize light scattering.[13]
Autofluorescence can occur in certain cell structures where the natural compound in the structure would fluoresce after being excited at relatively shorter wavelengths (similar to that of the excitation wavelength).[18] Induced fluorescence can also occur when certain non-autofluorescent compounds become fluorescent after binding to certain chemicals (such as formaldehyde).[13] These fluorescence can result in artifacts or background noise in the image. Noise from other fluorescence compounds can be effectively eliminated by using filters to capture the desired fluorescence wavelength, or by making sure the autofluorescence compounds are not present in the sample.
Current and future work
Modern fluorescence techniques attempt to incorporate methods to eliminate some blurring and noises. Optical aberrations are generally deterministic (it is constant throughout the image process and across different samples).[18] Deterministic blurring can be eliminated by deconvoluting the signal and subtracting the known artifact. The deconvolution technique is simply using an inverse fourier transform to obtain the original fluorescence signal and remove the artifact.[19]
Nevertheless, deconvolution has only been shown to work if there is a strong fluorescence signal or when the noise is clearly identified. In addition, deconvolution performs poorly because it does not include statistical information and can not reduce non-deterministic noise such as poissonian noise. To obtain better image resolution and quality, researchers have used statistical techniques to model the probability where photons may be distributed on the detector.[18][22] This technique, called the maximum likelihood method, is being further improved by algorithms to improve its performance speed.[22]
References
- 1 2 3 Fish, Kenneth N. (October 2009). "Total Internal Reflection Fluorescence (TIRF) Microscopy". Current Protocols in Cytometry. 0 12: Unit12.18. doi:10.1002/0471142956.cy1218s50. ISSN 1934-9297. PMC 4540339. PMID 19816922.
- 1 2 3 4 5 "Total Internal Reflection Fluorescence (TIRF) Microscopy". Nikon’s MicroscopyU. Retrieved 2021-12-06.
- 1 2 "What Is Total Internal Reflection Fluorescence (TIRF) Microscopy & Is It Right For You?". Cheeky Scientist. 2021-03-11. Retrieved 2021-12-06.
- ↑ Ambrose, EJ (24 Nov 1956). "A surface contact microscope for the study of cell movements". Nature. 178 (4543): 1194. Bibcode:1956Natur.178.1194A. doi:10.1038/1781194a0. PMID 13387666. S2CID 4290898.
- ↑ Axelrod, D. (1 April 1981). "Cell-substrate contacts illuminated by total internal reflection fluorescence". The Journal of Cell Biology. 89 (1): 141–145. doi:10.1083/jcb.89.1.141. PMC 2111781. PMID 7014571.
- ↑ Yanagida, Toshio; Sako, Yasushi; Minoghchi, Shigeru (10 February 2000). "Single-molecule imaging of EGFR signalling on the surface of living cells". Nature Cell Biology. 2 (3): 168–172. doi:10.1038/35004044. PMID 10707088. S2CID 25515586.
- ↑ Andre et al. Cross-correlated tirf/afm reveals asymmetric distribution of forcegenerating heads along self-assembled, synthetic myosin filaments. Biophysical Journal, 96:1952–1960, 2009.
- ↑ Sapkota, K.; et al. (2019). "Single-Step FRET-Based Detection of Femtomoles DNA". Sensors. 19 (16): 3495. Bibcode:2019Senso..19.3495S. doi:10.3390/s19163495. PMC 6719117. PMID 31405068.
- ↑ Ambrose, W; et al. (1999). "Single-molecule detection with total internal reflection excitation: comparing signal-to-background and total signals in different geometries". Cytometry. 36 (3): 224–31. doi:10.1002/(sici)1097-0320(19990701)36:3<224::aid-cyto12>3.0.co;2-j. PMID 10404972. S2CID 26302328.
- ↑ Mattheyses A. and Axelrod, D. (2006). "Direct measurement of the evanescent field profile produced by objective-based TIRF". J Biomed Opt. 11 (1): 014006A. doi:10.1117/1.2161018. PMID 16526883. S2CID 27507504.
- ↑ Brunstein M, Teremetz M, Hérault K, Tourain C, Oheim, M. (2014). "Eliminating unwanted far-field excitation in objective-type TIRF. Part I." Biophys. J. 106 (5): 1020–1032. Bibcode:2014BpJ...106.1020B. doi:10.1016/j.bpj.2013.12.049. PMC 4026778. PMID 24606927.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - 1 2 Microscopy: Total Internal Reflection Fluorescence (TIRF) Microscopy (Daniel Axelrod), retrieved 2021-12-06
- 1 2 3 4 "Contrast Techniques in Light Microscopy". Routledge & CRC Press. Retrieved 2021-12-06.
- 1 2 "Specialized Microscopy Techniques - Total Internal Reflection Fluorescence Microscopy | Olympus LS". www.olympus-lifescience.com. Retrieved 2021-12-06.
- ↑ "Fluorescence Microscopy - Fluorescence Filters | Olympus LS". www.olympus-lifescience.com. Retrieved 2021-12-06.
- ↑ Mueller, Michiel (2005-12-07). Introduction to Confocal Fluorescence Microscopy, Second Edition. doi:10.1117/3.639736. ISBN 9780819481177.
- ↑ K.K, Hamamatsu Photonics. "A visual guide to CCD vs. EM-CCD vs. CMOS". camera.hamamatsu.com. Retrieved 2021-12-06.
- 1 2 3 4 5 Mondal, Partha Pratim; Diaspro, Alberto (2014), Mondal, Partha Pratim; Diaspro, Alberto (eds.), "Image Reconstruction for Fluorescence Microscopy", Fundamentals of Fluorescence Microscopy: Exploring Life with Light, Dordrecht: Springer Netherlands, pp. 189–202, doi:10.1007/978-94-007-7545-9_10, ISBN 978-94-007-7545-9, retrieved 2021-12-06
- 1 2 Mueller, Michiel (2005-12-07). Introduction to Confocal Fluorescence Microscopy, Second Edition. doi:10.1117/3.639736. ISBN 9780819481177.
- ↑ O’Mara, P.; Farrell, A.; Bones, J.; Twomey, K. (2018-01-01). "Staying alive! Sensors used for monitoring cell health in bioreactors". Talanta. 176: 130–139. doi:10.1016/j.talanta.2017.07.088. hdl:10468/6193. ISSN 0039-9140. PMID 28917732.
- ↑ "Fluorophore Photobleaching Literature References". Nikon’s MicroscopyU. Retrieved 2021-12-06.
- 1 2 Varma, Mahesh Ravi; Rajan, K.; Mondal, Partha Pratim (2012-09-01). "Fast image reconstruction for fluorescence microscopy". AIP Advances. 2 (3): 032174. Bibcode:2012AIPA....2c2174V. doi:10.1063/1.4754604. S2CID 98034102.
- Axelrod, Daniel (1 November 2001). "Total Internal Reflection Fluorescence Microscopy in Cell Biology" (PDF). Traffic. 2 (11): 764–774. doi:10.1034/j.1600-0854.2001.21104.x. hdl:2027.42/72779. PMID 11733042. S2CID 15202097.
External links
- Interactive Fluorescence Dye and Filter Database Carl Zeiss Interactive Fluorescence Dye and Filter Database.
- TIRF Microscopy: Introduction and Applications TIRF Tutorial from Microscopy U
- TIRF Microscopy: Overview TIRF Tutorial from Olympus Microscopy Resource Center
- Olympus TIRFM Microscopes commercial TIRF microscope systems
- Carl Zeiss Laser TIRF 3 commercial TIRF microscope systems
- Lightguide- and prism-based TIRF microscopy TIRF-Labs.com :Commercial TIRF Microscopy and Spectroscopy. Selecting TIRFM geometry for your application
- TIRF FLIM microscopy Lambert Instruments TIRF - FLIM microscopy
- Schwartz Research Group, CU-Boulder Single Molecule Imaging Research Group
| Illumination and contrast methods |  | |
|---|---|---|
| Fluorescence methods | ||
| Sub-diffraction limit techniques | ||
| ||