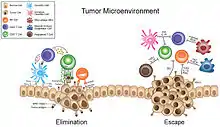
The tumor microenvironment (TME) is a complex ecosystem surrounding a tumor, composed of a variety of non-cancerous cells including blood vessels, immune cells, fibroblasts, signaling molecules and the extracellular matrix (ECM).[1][2][3][4] Mutual interaction between cancer cells and the different components of the TME support its growth and invasion in healthy tissues which correlates with tumor resistance to current treatments and poor prognosis. Tumors can influence the microenvironment by releasing extracellular signals, promoting tumor angiogenesis and inducing peripheral immune tolerance, while the immune cells in the microenvironment can affect the growth and evolution of cancerous cells.[1][5][6][7]
History
The importance of a stromal microenvironment, especially "wound" or regenerating tissue, has been recognized since the late 1800s. The interplay between the tumor and its microenvironment was part of Stephen Paget's 1889 "seed and soil" theory, in which he postulated that metastases of a particular type of cancer ("the seed") often metastasizes to certain sites ("the soil") based on the similarity of the original and secondary tumor sites.[8]
Its role in blunting an immune attack awaited the discovery of adaptive cellular immunity. In 1960, Klein and colleagues found that in mice, primary methylcholanthrene-induced sarcomas exhibited an antitumor immune response mediated by lymph node cells to cancer cells derived from the primary tumor. This immune response did not however affect the primary tumor. The primary tumor instead established a microenvironment that is functionally analogous to that of certain normal tissues, such as the eye.[3]
Later, mice experiments by Halachmi and Witz showed that for the same cancer cell line, greater tumorigenicity was evident in vivo than the same strain inoculated in vitro.[9][10]
Unambiguous evidence for the inability in humans of a systemic immune response to eliminate immunogenic cancer cells was provided by Boon's 1991 studies of antigens that elicit specific CD8+ T cell responses in melanoma patients. One such antigen was MAGE-A1. The coexistence of a progressing melanoma with melanoma-specific T cells implicitly does not involve immunoediting, but does not exclude the possibility of TME immune suppression.[3]
The discovery of melanoma-specific T cells in patients led to the strategy of adoptively transferring large numbers of in vitro-expanded tumor-infiltrating lymphocytes (TILs) which has proven that the immune system has the potential to control cancer. However, adoptive T cell therapy (ACT) with TILs has not had the dramatic success of ACT with virus-specific CD8+ T cells. The TME of solid cancers appears to be fundamentally different from that of the leukemias, in which clinical ACT trials with chimeric antigen receptor T cells have demonstrated efficacy.[3]
Vasculature
80–90% of cancer are carcinomas, or cancers that form from epithelial tissue.[11] This tissue is not vascularized, which prevents tumors from growing greater than 2mm in diameter without inducing new blood vessels.[12] Angiogenesis is upregulated to feed the cancer cells, and as a result the vasculature formed differs from that of normal tissue.
Enhanced permeability and retention effect
The enhanced permeability and retention effect (EPR) is the observation that the vasculature of tumors is often leaky and accumulates molecules in the blood stream to a greater extent than in normal tissue. This inflammation effect is not only seen in tumors, but in hypoxic areas of cardiac muscles following a myocardial infarction.[13][14] This permeable vasculature is thought to have several causes, including insufficient pericytes and a malformed basement membrane.[14]
Hypoxia
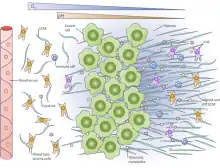
The tumor microenvironment is often hypoxic. As the tumor mass increases, the interior of the tumor becomes farther away from existing blood supply. While angiogenesis can reduce this effect, the partial pressure of oxygen is below 5 mm Hg (venous blood has a partial pressure of oxygen at 40 mm Hg) in more than 50% of locally advanced solid tumors.[15][16] The hypoxic environment leads to genetic instability, which is associated with cancer progression, via downregulating DNA repair mechanisms such as nucleotide excision repair (NER) and mismatch repair (MMR) pathways.[17] Hypoxia also causes the upregulation of hypoxia-inducible factor 1 alpha (HIF1-α), which induces angiogenesis and is associated with poorer prognosis and the activation of genes associated with metastasis,[16] leading, for instance, to increased cell migration and also ECM remodeling.[4]
While a lack of oxygen can cause glycolytic behavior in cells, some tumor cells also undergo aerobic glycolysis, in which they preferentially produce lactate from glucose even given abundant oxygen, called the Warburg effect.[18] No matter the cause, this leaves the extracellular microenvironment acidic (pH 6.5–6.9), while the cancer cells themselves are able to remain neutral (ph 7.2–7.4).[19] It has been shown that this induces greater cell migration in vivo and in vitro, possibly by promoting degradation of the ECM.[20][21]
Stromal cells
In cancer biology, the stroma is defined as the nonmalignant cells which are present in the tumor microenvironment. The stroma comprises a variable portion of the entire tumor; up to 90% of a tumor may be stroma, with the remaining 10% as cancer cells. Many types of cells are present in the stroma, but four abundant types are fibroblasts, T cells, macrophages, and endothelial cells.[22] The stroma surrounding a tumor often reacts to intrusion via inflammation, similar to how it might respond to a wound.[23] Inflammation can encourage angiogenesis, speed the cell cycle and prevent cell death, all of which augments tumor growth.[24]
Carcinoma associated fibroblasts
Carcinoma associated fibroblasts (CAFs) are a heterogeneous group of fibroblasts whose function is pirated by cancer cells and redirected toward carcinogenesis.[25] These cells are usually derived from the normal fibroblasts in the surrounding stroma but can also come from pericytes, smooth muscle cells, fibrocytes, mesenchymal stem cells (MSCs, often derived from bone marrow), or via epithelial-mesenchymal transition (EMT) or endothelial-mesenchymal transition (EndMT).[26][27][28] Unlike their normal counterparts, CAFs do not retard cancer growth in vitro.[29] CAFs perform several functions that support tumor growth, such as secreting vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF), and other pro-angiogenic signals to induce angiogenesis.[15] CAFs can also secrete transforming growth factor beta (TGF-β), which is associated with EMT, a process by which cancer cells can metastasize,[30] and is associated with inhibiting cytotoxic T cells and natural killer T cells.[31] As fibroblasts, CAFs are able to rework the ECM to include more paracrine survival signals such as IGF-1 and IGF-2, thus promoting survival of the surrounding cancer cells. CAFs are also associated with the Reverse Warburg Effect where the CAFs perform aerobic glycolysis and feed lactate to the cancer cells.[25]
Several markers identify CAFs, including expression of α smooth muscle actin (αSMA), vimentin, platelet-derived growth factor receptor α (PDGFR-α), platelet-derived growth factor receptor β (PDGFR-β), fibroblast specific protein 1 (FSP-1) and fibroblast activation protein (FAP).[27] None of these factors can be used to differentiate CAFs from all other cells by itself.
Extracellular matrix remodeling

Fibroblasts are in charge of laying down most of the collagens, elastin, glycosaminoglycans, proteoglycans (e.g. perlecan), and glycoproteins in the ECM. As many fibroblasts are transformed into CAFs during carcinogenesis, this reduces the amount of ECM produced and the ECM that is produced can be malformed, like collagen being loosely woven and non-planar, possibly even curved.[32] In addition, CAFs produce matrix metalloproteinases (MMP) that cleave the proteins within the ECM.[15] CAFs are also able to disrupt the ECM via force, generating a track that a carcinoma cell can follow.[33] In either case, destruction of the ECM allows cancer cells to escape from their in situ location and intravasate into the blood stream where they can metastasize systematically. It can also provide passage for endothelial cells to complete angiogenesis to the tumor site.
Destruction of the ECM also modulates the signaling cascades controlled by the interaction of cell-surface receptors and the ECM, and it also reveals binding sites previously hidden, like the integrin alpha-v beta-3 (αVβ3) on the surface of melanoma cells can be ligated to rescue the cells from apoptosis after degradation of collagen.[34][35] In addition, the degradation products may have downstream effects as well that can increase cancer cell tumorigenicity and can serve as potential biomarkers.[34] ECM destruction also releases the cytokines and growth factors stored therein (for example, VEGF, basic fibroblast growth factor (bFGF), insulin-like growth factors (IGF1 and IGF2), TGF-β, EGF, heparin-binding EGF-like growth factor (HB-EGF), and tumor necrosis factor (TNF), which can increase the growth of the tumor.[32][36] Cleavage of ECM components can also release cytokines that inhibit tumorigenesis, such as degradation of certain types of collagen can form endostatin, restin, canstatin and tumstatin, which have antiangiogenic functions.[32]
ECM stiffening is associated with tumor progression.[4][37] This stiffening may be partially attributed to CAFs secreting lysyl oxidase (LOX), an enzyme that cross-links the collagen IV found in the ECM.[27][38]
Immune cells
_of_breast_cancer_models.svg.png.webp)

Myeloid-derived suppressor cells and tumor associated macrophages
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells of myelogenous origin with the potential to repress T cell responses. They regulate wound repair and inflammation and are rapidly expanded in cancer, correlating with that signs of inflammation are seen in most if not all tumor sites.[39][40] Tumors can produce exosomes that stimulate inflammation via MDSCs.[41][42] This group of cells include some tumor associated macrophages (TAMs).[39] TAMs are a central component in the strong link between chronic inflammation and cancer. TAMs are recruited to the tumor as a response to cancer-associated inflammation.[43] Unlike normal macrophages, TAMs lack cytotoxic activity.[44] TAMs have been induced in vitro by exposing macrophage progenitors to different immune regulatory cytokines, such as interleukin 4 (IL-4) and interleukin 13 (IL-13).[25] TAMs gather in necrotic regions of tumors where they are associated with hiding cancer cells from normal immune cells by secreting interleukin 10 (IL-10), aiding angiogenesis by secreting vascular endothelial growth factor (VEGF) and nitric oxide synthase(NOS),[15] supporting tumor growth by secreting epidermal growth factor (EGF)[45] and remodeling the ECM.[15] TAMs show sluggish NF-κB activation, which allows for the smoldering inflammation seen in cancer.[46] An increased amount of TAMs is associated with worse prognosis.[47][48] TAMs represent a potential target for novel cancer therapies.
TAMs are associated with using exosomes to deliver invasion-potentiating microRNA (miRNA) into cancerous cells, specifically breast cancer cells.[41][49]
Neutrophils
Neutrophils are polymorphonuclear immune cells that are critical components of the innate immune system. Neutrophils can accumulate in tumors and in some cancers, such as lung adenocarcinoma, their abundance at the tumor site is associated with worsened disease prognosis.[50][51][52] When compared among 22 different tumor infiltrating leukocyte (TIL) subsets, neutrophils are especially important predictors of survival for patients with solid tumors.[51] Neutrophil numbers (and myeloid cell precursors) in the blood can be increased in some patients with solid tumors.[53][54][55] Experiments in mice have mainly shown that tumor-associated neutrophils exhibit tumor-promoting functions,[56][57][58][59][60][61] but a smaller number of studies show that neutrophils can also inhibit tumor growth.[62][63] Neutrophil phenotypes are diverse and distinct neutrophil phenotypes in tumors have been identified.[64][58] In mice, neutrophils and 'granulocytic myeloid derived suppressor cells' are often identified by the same cell surface antibodies using flow cytometry and it is unclear whether these are overlapping or distinct populations.[65][66]
Tumor infiltrating lymphocytes
Tumor infiltrating lymphocytes (TILs) are lymphocytes that penetrate a tumor. TILs have a common origin with myelogenous cells at the hematopoietic stem cell, but diverge in development. Concentration is generally positively correlated.[45] However, only in melanoma has autologous TIL transplant succeeded as a treatment.[67] Cancer cells induce apoptosis of activated T cells (a class of lymphocyte) by secreting exosomes containing death ligands such as FasL and TRAIL, and via the same method, turn off the normal cytotoxic response of natural killer cells (NK cells).[42][68] This suggests that cancer cells actively work to restrain TILs.
T cells
Preclinical mice studies implicate CAFs, TAMs and myelomonocytic cells (including several myeloid-derived suppressor cells (MDSCs)) in restricting T cell accumulation near cancer cells. Overcoming this restriction, combined with a T cell checkpoint antagonist, revealed enhanced antitumor effects. Tumor vasculature also plays an active role in restricting T cell entry into the TME.[3]
T cells reach tumor sites via the circulatory system. The TME appears to preferentially recruit other immune cells over T cells from that system. One such mechanism is the release of cell-type specific chemokines. Another is the TME's capacity to posttranslationally alter chemokines. For example, the production of reactive nitrogen species by MDSCs within the TME induces nitration of CCL2 (N-CCL2), which traps T cells in the stroma of colon and prostate cancers. N-CCL2 does attract monocytes. CCL2 nitration inhibitors enhanced the accumulation of TILs in the corresponding animal models and resulted in improved efficacy of ACT.[3]
Another T cell inhibitor appears to be the apoptosis inducer Fas ligand (FasL) that is found in the tumor vasculature of tumor types including ovarian, colon, prostate, breast, bladder and renal cancer. High levels of endothelial FasL are accompanied by few CD8+ T cells, but abundant regulatory T cells (Tregs). In preclinical models inhibiting FasL increased the ratio of tumor-rejecting T cells to Treg cells and T cell–dependent tumor suppression. FasL inhibition also improves ACT efficacy.[3] For many cancers, an increased frequency of in the tumor microenvironment is associated with worse outcomes for the individual. This is not the case with colorectal cancer; an increased frequency of Treg cells may suppress inflammation mediated by the gut flora, which promotes tumor growth.[69]
In ovarian cancer elevated VEGF levels and expression of the immune regulatory ligand B7H3 (CD276), or the endothelin B receptor (ETBR) on tumor vessels correlate with decreased T cell infiltration and worse clinical outcome. Pharmacological inhibition of ETBR increased T cell adhesion to endothelial cells in an intercellular adhesion molecule-1 (ICAM-1)–dependent manner, increasing TIL numbers in mice and a corresponding tumor response. Anti-angiogenic inhibitors targeting VEGF and its receptor VEGFR2 (approved for treatment of multiple cancers) induce vascular normalization. This, in turn, increases TILs and improves ACT and vaccine efficacy in preclinical models. VEGF impairs DC maturation, offering another means to enhance intratumoral immune responses. Deleting the regulator of G-protein signaling, Rgs5 reduced vessel leakiness and hypoxia, enhanced T cell infiltration into mouse pancreatic neuroendocrine tumors, and prolonged animal survival. Vascular normalization is thus likely more effective than vessel destruction. Targeted delivery of tumor necrosis factor-α (TNF-α) was reported to normalize tumor blood vessels, increase CD8+ T cell infiltration and enhance vaccine and ACT therapies, unlike inflammatory cytokines interferon-γ (IFN-γ).[3]
Reproduction
T cells must reproduce after arriving at the tumor site to further increase their numbers, survive the TME's hostile elements and migrate through the stroma to the cancer cells. The TME obstructs all three activities. The draining lymph nodes are the likely location for T cell clonal reproduction, although this also occurs within the tumor. Preclinical models suggest that the TME is the major site of cancer-specific T cell cloning and that the CD8+ T cell replicative response there is orchestrated by the CD103+, Baft3-dependent DC, which can efficiently cross-present cancer cell antigens, suggesting that therapeutic interventions that enhance CD103+ contribute to tumor control. Among such strategies are antibodies to the interleukin-10 receptor (IL10R). In a mammary carcinoma mouse model it neutralized the effects of TAM-produced IL10, relieved the suppression of IL12 production by intratumoral DCs and improved the CD8+ T cell–dependent antitumor effects of chemotherapy. A similar outcome was achieved by neutralizing macrophage colony-stimulating factor 1, which impaired the intratumoral accumulation of TAMs. Another strategy is the administration of antibody-interferon-β (IFN-β) complexes that activate intratumoral DCs to cross-present antigen to CD8+ T cells. They are targeted against oncogenic receptors such as epidermal growth factor receptor (EGFR).[3]
Tumor eradication resulted when PD-L1 (also induced by IFN-β acting on DCs) was neutralized. DC function also may be adversely affected by the TME's hypoxic conditions, which induces PD-L1 expression on DCs and other myelomonocytic cells as a result of hypoxia-inducible factors-1α (HIF-1α) binding directly to a hypoxia-responsive element in the PD-L1 promoter. Even the aerobic glycolysis of cancer cells may antagonize local immune reactions via increasing lactate production, which induces the M2 TAM polarization. An M1 to M2 phenotypic transition of intratumoral macrophages was reported after the induction of cancer cell apoptosis in human and mouse gastrointestinal stromal tumors by KIT oncoprotein inhibitor imatinib. The designation of M1 and M2 polarization states over-simplify macrophage biology, since at least six different TAM subpopulations are known. Therefore, TME TAM phenotype descriptors are likely important.[3]
The TME may also directly impair intratumoral T cell proliferation. Indole 2,3-dioxygenase (IDO)—which can be expressed by DCs, MDSCs and cancer cells—catabolizes tryptophan and generates kynurenine. Both the deprivation of tryptophan and the generation of its metabolic product inhibit clonal T cell expansion. IDO also promotes the conversion of T cells to Treg cells and increases IL-6 expression, which augments MDSC functions. Accordingly, IDO1 genetic deficiency is associated with reduced tumor burden and metastasis and enhanced survival in mouse models of lung and breast cancer. The therapeutic potential of inhibiting IDO, in combination with anti-CTLA-4 was demonstrated in the B16 melanoma model and was associated with increased intratumoral T cells. IDO's capacity to block Treg cell to helperlike cell reprogramming by sustaining transcription factor Eos and the transcriptional program it regulates, also suppresses the immune response.[3]
Apoptosis
The TME can limit T cell viability. Both IDO and PD-L1 may induce T cell apoptosis. Myelomonocytic cell products that cause apoptosis include FasL, TNF-α, and TNF-related apoptosis-inducing ligand (TRAIL). Ppp2r2d is a key regulator promoting T cell apoptosis and suppressing T cell proliferation.[3]
TAMs and MDSCs
Targeting intratumoral TAMs and MDSCs can also reduce tumor burdens in preclinical models, in both T cell–dependent and T cell–independent ways. For instance, inhibiting chemokine receptor type 2 (CCR2), colony-stimulating factor-1 receptor (CSF-1R) and granulocyte macrophage colony-stimulating factor (GM-CSF) in preclinical models of melanoma, pancreatic, breast, and prostatic carcinoma increased T cells and restricted tumor growth. The effect was enhanced by anti-CTLA-4 or anti-PD-1/PD-L1. These studies did not determine whether the increases in T cells were a consequence of viability or replication.[3]
Inhibition of CSF-1R in a preclinical proneural glioblastoma multiforme model and in patient-derived glioma xenografts increased survival and shrank established tumors in an apparently T cell–independent manner that correlated with the reprogramming of macrophages away from an M2 phenotype. Similarly, an activator of TAMs, an agonistic antibody to CD40, when administered in combination with the chemotherapeutic drug gemcitabine, suppressed mouse PDA growth in a T cell–independent manner, suggesting that stimulated macrophages may have anticancer functions.[3]
B cells regulate TAM phenotypes in squamous cell carcinoma TME. Correspondingly, B cell depletion reprogrammed TAMs, thus reducing their suppression of CD8 cells and enhancing chemotherapy. An autochthonous melanoma mouse model depleted Treg cells and neutralized IL-10, revealing tumor-killing properties. TAMs mediate the effects of antitumor antibodies and genetically engineered ligands that interact with CD47 to prevent the CD47/signal regulatory protein–α (SIRPα) signaling system from suppressing antibody-coated cancer cell phagocytosis.[3]
Spatial distribution
CAFs restrict T cell distribution by two means. They can physically exclude them, as mediated by their extracellular matrix. T cell motility was higher in regions of loose fibronectin and collagen than in dense matrix areas surrounding tumor nests. Collagenase added to reduce matrix rigidity or chemokine CCL5 experimentally produced by tumor cells increased movement into contact with cancer cells.
They can also exclude them via biosynthesis of CXCL12. Conditionally depleting these cells from the stroma of an ectopic, transplanted tumor and of an autochthonous pancreatic ductal adenocarcinoma (PDA) allowed T cells to rapidly control tumor growth. However, the depletion must be limited to the TME, because these cells carry out essential functions in several normal tissues. "Reprogramming" FAP+ cells in the TME with a vitamin D analog may neutralize them. Another approach may block their immune suppressive mechanism. In a preclinical PDA mouse model, FAP+ CAFs produced the chemokine CXCL12, which is bound by PDA cancer cells. Because FAP+ stromal cells also accumulate in nontransformed, inflammatory lesions, this "coating" of cancer cells may reflect a means by which "injured" epithelial cells protect themselves from immune attack. Administering an inhibitor of CXCL12 receptor CXCR4 caused the rapid spread of T cells among cancer cells, arrested tumor growth and stimulated tumor sensitivity to anti-PD-L1.
Research and Clinical implications
Models of the tumor microenvironment
Several in vitro and in vivo models have been developed that seek to replicate the TME in a controlled environment. Tumor immortalised cell lines and primary cell cultures have been long used in order to study various tumors. They are quick to set up and inexpensive, but simplistic and prone to genetic drift.[70] 3D tumor models have been developed as a more spatially representative model of the TME. Spheroid cultures, scaffolds and organoids are generally derived from stem cells or ex vivo and are much better at recreating the tumour architecture than 2D cell cultures.[71] Recent advancement is the use of microfluidic platforms, also called tumor-on-a-chip platforms, in investigating cancer-immune crosstalk.[72][73] These devices can be used to recapitulate the TME allowing broader understanding of specific interactions of cancer cells and the surrounding environment, as well as assess the efficacy of different immunotherapies available.[73]
In vivo models induce tumours in animals allowing for a more systematic study of the TME, but problems arise in the translation of human proteins in animal tissue as well as the immune interaction of different species. Early models relied on carcinogen induction and transplantation of tumours, while today experiments can also include genetically engineered animals.[7] Humanized mouse models offer the most comparable TME to human patients, but are time and resource intensive systems to maintain.[74][75]
Drug development
High throughput cancer therapeutics screens are performed in vitro without the accompanying microenvironment. However, studies also investigate the effects of supportive stroma cells and their resistance to therapy.[1] The latter studies revealed interesting therapeutic targets in the microenvironment including integrins and chemokines. These were missed by initial screens for anti-cancer drugs and might also help explain why so few drugs are highly potent in vivo.
Advances in TME remodeling nanotherapeutics suppress cancer metastasis and recurrence.[76] Numerous strategies employing nanotechnology to control TAM polarization have been created and examined. Researchers have discovered that the use of ferumoxytol suppress tumor growth by inducing transition of M2 macrophage to pro-inflammatory M1 phenotype.[77] Nanocarrier vehicles (~20–200 nm in diameter) can transport drugs and other therapeutic molecules. These therapies can be targeted to selectively extravasate through tumor vasculature via the EPR effect. Nanocarriers are now considered the gold standard of targeted cancer therapy because it can target tumors that are hypovascularized, such as prostate and pancreatic tumors.[14][78] These efforts include protein capsids[79] and liposomes.[80] However, as some important, normal tissues, such as the liver and kidneys, also have fenestrated endothelium, the nanocarrier size (10–100 nm, with greater retention in tumors seen in using larger nanocarriers) and charge (anionic or neutral) must be considered.[14] Lymphatic vessels do not usually develop with the tumor, leading to increased interstitial fluid pressure, which may block tumor access.[14][81]
Therapies
Antibodies
Bevacizumab is clinically approved in the US to treat a variety of cancers by targeting VEGF-A, which is produced by both CAFs and TAMs, thus slowing angiogenesis.
Targeting immunoregulatory membrane receptors succeeded in some patients with melanoma, non-small-cell lung carcinoma, urothelial bladder cancer and renal cell cancer. In mice, anti-CTLA-4 therapy leads to clearance from the tumor of Foxp3+ regulatory T cells (Treg cells) whose presence may impair effector T cell function.[82] Similarly anti-PD-1/anti-PD-L1 therapy blocks the inhibitory PD-1 receptor. Other, potentially more fundamental TME inhibitory reactions (as in microsatellite stable colorectal cancer, ovarian cancer, prostate cancer, and PDA have yet to be overcome. The TME appears to aid in excluding killer T cells from the vicinity of cancer cells.[3]
Kinase inhibitors
Many other small molecule kinase inhibitors block the receptors for the growth factors released, thus making the cancer cell deaf to much of the paracrine signaling produced by CAFs and TAMs. These inhibitors include sunitinib, pazopanib, sorafenib and axitinib, all of which inhibit platelet derived growth factor receptors (PDGF-Rs) and VEGF receptors (VEGFRs). Cannabidiol (a cannabis derivate without psychoactive effects) has also been shown to inhibit the expression of VEGF in Kaposi's sarcoma.[83] Natalizumab is a monoclonal antibody that targets a molecule responsible for cell adhesion (integrin VLA-4) and has promising in vitro activity in B cell lymphomas and leukemias.
Trabectedin has immunomodulatory effects that inhibit TAMs.[45]
Liposomes
Liposome formulations encapsulate anti-cancer drugs for selective uptake to tumors via the EPR effect. They are included Doxil and Myocet, both of which encapsulate doxorubicin (a DNA intercalator and common chemotherapeutic); DaunoXome, which encapsulates daunorubicin (a similar DNA intercalator); and Onco-TCS, which encapsulates vincristine (a molecule that induces formation of microtubules, dysregulating cell division). Another novel utilization of the EPR effect comes from Protein-bound paclitaxel (marketed under the trade name Abraxane) where paclitaxel is bound to albumin to add bulk and aid delivery.
CAR T cell therapy
Chimeric antigen receptors (CAR) T cell therapy is an immunotherapy treatment that uses genetically modified T lymphocytes to effectively target tumor cells.[84][85] Since the TME is known for several barriers that limits the ability of CAR T cells to infiltrate the tumor, several strategies have been developed to address this. Localized delivery of CAR T cells in glioblastoma suggested improved anti-tumor activity and engineering these cells to overexpress chemokine receptors suggested improvement of CAR T cell trafficking to the TME.[86][87][88]
See also
References
- 1 2 3 Alfarouk KO, Muddathir AK, Shayoub ME (January 2011). "Tumor acidity as evolutionary spite". Cancers. 3 (1): 408–414. doi:10.3390/cancers3010408. PMC 3756368. PMID 24310355.
- ↑ "NCI Dictionary of Cancer Terms". National Cancer Institute. 2011-02-02.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Joyce JA, Fearon DT (April 2015). "T cell exclusion, immune privilege, and the tumor microenvironment". Science. 348 (6230): 74–80. Bibcode:2015Sci...348...74J. doi:10.1126/science.aaa6204. PMID 25838376.
- 1 2 3 Spill F, Reynolds DS, Kamm RD, Zaman MH (August 2016). "Impact of the physical microenvironment on tumor progression and metastasis". Current Opinion in Biotechnology. 40: 41–48. doi:10.1016/j.copbio.2016.02.007. PMC 4975620. PMID 26938687.
- ↑ Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA (January 2017). "TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis". Cytokine. 89: 127–135. doi:10.1016/j.cyto.2016.01.021. PMID 26854213.
- ↑ Ghoshdastider U, Rohatgi N, Mojtabavi Naeini M, Baruah P, Revkov E, Guo YA, et al. (April 2021). "Pan-Cancer Analysis of Ligand-Receptor Cross-talk in the Tumor Microenvironment". Cancer Research. 81 (7): 1802–1812. doi:10.1158/0008-5472.CAN-20-2352. PMID 33547160. S2CID 232432582.
- 1 2 Žavbi J, Breznik B (2021). "Modelling the microenvironment of the most aggressive brain tumours for preclinical studies". Advances in Cancer Biology - Metastasis. 3: 100017. doi:10.1016/j.adcanc.2021.100017. ISSN 2667-3940. S2CID 244452599.
- ↑ The Lancet, Volume 133, Issue 3421, 23 March 1889, Pages 571-573
- ↑ Halachmi E, Witz IP (May 1989). "Differential tumorigenicity of 3T3 cells transformed in vitro with polyoma virus and in vivo selection for high tumorigenicity" (PDF). Cancer Research. 49 (9): 2383–2389. PMID 2539901.
- ↑ Witz IP, Levy-Nissenbaum O (October 2006). "The tumor microenvironment in the post-PAGET era". Cancer Letters. 242 (1): 1–10. doi:10.1016/j.canlet.2005.12.005. PMID 16413116.
- ↑ "Cancer Overview". Standford Medicine Cancer Institute.
- ↑ Duffy MJ (1996). "The biochemistry of metastasis". Advances in Clinical Chemistry. 32: 135–166. doi:10.1016/S0065-2423(08)60427-8. ISBN 9780120103324. PMID 8899072.
- ↑ Palmer TN, Caride VJ, Caldecourt MA, Twickler J, Abdullah V (March 1984). "The mechanism of liposome accumulation in infarction". Biochimica et Biophysica Acta (BBA) - General Subjects. 797 (3): 363–368. doi:10.1016/0304-4165(84)90258-7. PMID 6365177.
- 1 2 3 4 5 Danhier F, Feron O, Préat V (December 2010). "To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery". Journal of Controlled Release. 148 (2): 135–146. doi:10.1016/j.jconrel.2010.08.027. PMID 20797419.
- 1 2 3 4 5 Weber CE, Kuo PC (September 2012). "The tumor microenvironment". Surgical Oncology. 21 (3): 172–177. doi:10.1016/j.suronc.2011.09.001. PMID 21963199.
- 1 2 Blagosklonny MV (January 2004). "Antiangiogenic therapy and tumor progression". Cancer Cell. 5 (1): 13–17. doi:10.1016/S1535-6108(03)00336-2. PMID 14749122.
- ↑ Bindra RS, Glazer PM (January 2005). "Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis". Mutation Research. 569 (1–2): 75–85. doi:10.1016/j.mrfmmm.2004.03.013. PMID 15603753.
- ↑ Gatenby RA, Gillies RJ (November 2004). "Why do cancers have high aerobic glycolysis?". Nature Reviews. Cancer. 4 (11): 891–899. doi:10.1038/nrc1478. PMID 15516961. S2CID 10866959.
- ↑ Lee SH, Griffiths JR (June 2020). "How and Why Are Cancers Acidic? Carbonic Anhydrase IX and the Homeostatic Control of Tumour Extracellular pH". Cancers. 12 (6): 1616. doi:10.3390/cancers12061616. PMC 7352839. PMID 32570870.
- ↑ van Sluis R, Bhujwalla ZM, Raghunand N, Ballesteros P, Alvarez J, Cerdán S, et al. (April 1999). "In vivo imaging of extracellular pH using 1H MRSI". Magnetic Resonance in Medicine. 41 (4): 743–750. doi:10.1002/(SICI)1522-2594(199904)41:4<743::AID-MRM13>3.0.CO;2-Z. PMID 10332850.
- ↑ Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. (March 2013). "Acidity generated by the tumor microenvironment drives local invasion". Cancer Research. 73 (5): 1524–1535. doi:10.1158/0008-5472.CAN-12-2796. PMC 3594450. PMID 23288510.
- ↑ Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW (July 1991). "Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts". Cancer Research. 51 (14): 3753–3761. PMID 1712249.
- ↑ Dvorak HF (December 1986). "Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing". The New England Journal of Medicine. 315 (26): 1650–1659. doi:10.1056/NEJM198612253152606. PMID 3537791.
- ↑ Kundu JK, Surh YJ (July–August 2008). "Inflammation: gearing the journey to cancer". Mutation Research. 659 (1–2): 15–30. doi:10.1016/j.mrrev.2008.03.002. PMID 18485806.
- 1 2 3 Hanahan D, Coussens LM (March 2012). "Accessories to the crime: functions of cells recruited to the tumor microenvironment". Cancer Cell. 21 (3): 309–322. doi:10.1016/j.ccr.2012.02.022. PMID 22439926.
- ↑ Räsänen K, Vaheri A (October 2010). "Activation of fibroblasts in cancer stroma". Experimental Cell Research. 316 (17): 2713–2722. doi:10.1016/j.yexcr.2010.04.032. PMID 20451516.
- 1 2 3 Marsh T, Pietras K, McAllister SS (July 2013). "Fibroblasts as architects of cancer pathogenesis". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1832 (7): 1070–1078. doi:10.1016/j.bbadis.2012.10.013. PMC 3775582. PMID 23123598.
- ↑ Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. (February 2011). "Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth". Cancer Cell. 19 (2): 257–272. doi:10.1016/j.ccr.2011.01.020. PMC 3060401. PMID 21316604.
- ↑ Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N, et al. (June 2011). "High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts". International Journal of Cancer. 128 (12): 2793–2802. doi:10.1002/ijc.25612. hdl:10616/40777. PMID 20715102. S2CID 27493689.
- ↑ Chaffer CL, Weinberg RA (March 2011). "A perspective on cancer cell metastasis". Science. 331 (6024): 1559–1564. Bibcode:2011Sci...331.1559C. doi:10.1126/science.1203543. PMID 21436443. S2CID 10550070.
- ↑ Stover DG, Bierie B, Moses HL (July 2007). "A delicate balance: TGF-beta and the tumor microenvironment". Journal of Cellular Biochemistry. 101 (4): 851–861. doi:10.1002/jcb.21149. PMID 17486574. S2CID 206014864.
- 1 2 3 Tlsty TD, Coussens LM (February 2006). "Tumor stroma and regulation of cancer development". Annual Review of Pathology. 1: 119–150. doi:10.1146/annurev.pathol.1.110304.100224. PMID 18039110.
- ↑ Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E (December 2007). "Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells". Nature Cell Biology. 9 (12): 1392–1400. doi:10.1038/ncb1658. PMID 18037882. S2CID 35445729.
- 1 2 Pupa SM, Ménard S, Forti S, Tagliabue E (September 2002). "New insights into the role of extracellular matrix during tumor onset and progression". Journal of Cellular Physiology. 192 (3): 259–267. doi:10.1002/jcp.10142. PMID 12124771. S2CID 31791792.
- ↑ Montgomery AM, Reisfeld RA, Cheresh DA (September 1994). "Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen". Proceedings of the National Academy of Sciences of the United States of America. 91 (19): 8856–8860. Bibcode:1994PNAS...91.8856M. doi:10.1073/pnas.91.19.8856. PMC 44705. PMID 7522323.
- ↑ Bergers G, Coussens LM (February 2000). "Extrinsic regulators of epithelial tumor progression: metalloproteinases". Current Opinion in Genetics & Development. 10 (1): 120–127. doi:10.1016/S0959-437X(99)00043-X. PMID 10679388.
- ↑ Sinkus R, Lorenzen J, Schrader D, Lorenzen M, Dargatz M, Holz D (June 2000). "High-resolution tensor MR elastography for breast tumour detection". Physics in Medicine and Biology. 45 (6): 1649–1664. Bibcode:2000PMB....45.1649S. doi:10.1088/0031-9155/45/6/317. PMID 10870716. S2CID 250815945.
- ↑ Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. (November 2009). "Matrix crosslinking forces tumor progression by enhancing integrin signaling". Cell. 139 (5): 891–906. doi:10.1016/j.cell.2009.10.027. PMC 2788004. PMID 19931152.
- 1 2 Bronte V, Grabrilovich D (2010). "Myeloid-derived suppressor cells (Poster)" (PDF). Nature.
- ↑ Mantovani A, Allavena P, Sica A, Balkwill F (July 2008). "Cancer-related inflammation". Nature. 454 (7203): 436–444. Bibcode:2008Natur.454..436M. doi:10.1038/nature07205. hdl:2434/145688. PMID 18650914. S2CID 4429118.
- 1 2 Mathias RA, Gopal SK, Simpson RJ (January 2013). "Contribution of cells undergoing epithelial-mesenchymal transition to the tumour microenvironment". Journal of Proteomics. 78: 545–557. doi:10.1016/j.jprot.2012.10.016. PMID 23099347.
- 1 2 Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L (April 2007). "Tumor-released microvesicles as vehicles of immunosuppression". Cancer Research. 67 (7): 2912–2915. doi:10.1158/0008-5472.CAN-07-0520. PMID 17409393.
- ↑ Balkwill F, Charles KA, Mantovani A (March 2005). "Smoldering and polarized inflammation in the initiation and promotion of malignant disease". Cancer Cell. 7 (3): 211–217. doi:10.1016/j.ccr.2005.02.013. PMID 15766659.
- ↑ Qian BZ, Pollard JW (April 2010). "Macrophage diversity enhances tumor progression and metastasis". Cell. 141 (1): 39–51. doi:10.1016/j.cell.2010.03.014. PMC 4994190. PMID 20371344.
- 1 2 3 Solinas G, Germano G, Mantovani A, Allavena P (November 2009). "Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation". Journal of Leukocyte Biology. 86 (5): 1065–1073. doi:10.1189/jlb.0609385. hdl:2318/1740263. PMID 19741157. S2CID 6573469.
- ↑ Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. (March 2006). "A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation)". Blood. 107 (5): 2112–2122. doi:10.1182/blood-2005-01-0428. PMID 16269622. S2CID 5884781.
- ↑ Zhang W, Wang L, Zhou D, Cui Q, Zhao D, Wu Y (January 2011). "Expression of tumor-associated macrophages and vascular endothelial growth factor correlates with poor prognosis of peripheral T-cell lymphoma, not otherwise specified". Leukemia & Lymphoma. 52 (1): 46–52. doi:10.3109/10428194.2010.529204. PMID 21077742. S2CID 26116121.
- ↑ Zhang BC, Gao J, Wang J, Rao ZG, Wang BC, Gao JF (December 2011). "Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma". Medical Oncology. 28 (4): 1447–1452. doi:10.1007/s12032-010-9638-5. PMID 20676804. S2CID 24840259.
- ↑ Yang M, Chen J, Su F, Yu B, Su F, Lin L, et al. (September 2011). "Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells". Molecular Cancer. 10 (117): 117. doi:10.1186/1476-4598-10-117. PMC 3190352. PMID 21939504.
- ↑ Coffelt SB, Wellenstein MD, de Visser KE (July 2016). "Neutrophils in cancer: neutral no more" (PDF). Nature Reviews. Cancer. 16 (7): 431–446. doi:10.1038/nrc.2016.52. PMID 27282249. S2CID 4393159.
- 1 2 Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. (August 2015). "The prognostic landscape of genes and infiltrating immune cells across human cancers". Nature Medicine. 21 (8): 938–945. doi:10.1038/nm.3909. PMC 4852857. PMID 26193342.
- ↑ Engblom C, Pfirschke C, Pittet MJ (July 2016). "The role of myeloid cells in cancer therapies". Nature Reviews. Cancer. 16 (7): 447–462. doi:10.1038/nrc.2016.54. PMID 27339708. S2CID 21924175.
- ↑ Huang SH, Waldron JN, Milosevic M, Shen X, Ringash J, Su J, et al. (February 2015). "Prognostic value of pretreatment circulating neutrophils, monocytes, and lymphocytes in oropharyngeal cancer stratified by human papillomavirus status". Cancer. 121 (4): 545–555. doi:10.1002/cncr.29100. PMID 25336438. S2CID 926930.
- ↑ Jiang L, Jiang S, Situ D, Lin Y, Yang H, Li Y, et al. (April 2015). "Prognostic value of monocyte and neutrophils to lymphocytes ratio in patients with metastatic soft tissue sarcoma". Oncotarget. 6 (11): 9542–9550. doi:10.18632/oncotarget.3283. PMC 4496237. PMID 25865224.
- ↑ Wu WC, Sun HW, Chen HT, Liang J, Yu XJ, Wu C, et al. (March 2014). "Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients". Proceedings of the National Academy of Sciences of the United States of America. 111 (11): 4221–4226. Bibcode:2014PNAS..111.4221W. doi:10.1073/pnas.1320753111. PMC 3964061. PMID 24591638.
- ↑ Faget J, Groeneveld S, Boivin G, Sankar M, Zangger N, Garcia M, et al. (December 2017). "Neutrophils and Snail Orchestrate the Establishment of a Pro-tumor Microenvironment in Lung Cancer". Cell Reports. 21 (11): 3190–3204. doi:10.1016/j.celrep.2017.11.052. PMID 29241546.
- ↑ Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. (June 2015). "IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis". Nature. 522 (7556): 345–348. Bibcode:2015Natur.522..345C. doi:10.1038/nature14282. PMC 4475637. PMID 25822788.
- 1 2 Engblom C, Pfirschke C, Zilionis R, Da Silva Martins J, Bos SA, Courties G, et al. (December 2017). "Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils". Science. 358 (6367): eaal5081. doi:10.1126/science.aal5081. PMC 6343476. PMID 29191879.
- ↑ Casbon AJ, Reynaud D, Park C, Khuc E, Gan DD, Schepers K, et al. (February 2015). "Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils". Proceedings of the National Academy of Sciences of the United States of America. 112 (6): E566–E575. Bibcode:2015PNAS..112E.566C. doi:10.1073/pnas.1424927112. PMC 4330753. PMID 25624500.
- ↑ Wculek SK, Malanchi I (December 2015). "Neutrophils support lung colonization of metastasis-initiating breast cancer cells". Nature. 528 (7582): 413–417. Bibcode:2015Natur.528..413W. doi:10.1038/nature16140. PMC 4700594. PMID 26649828.
- ↑ Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. (December 2010). "Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes". Proceedings of the National Academy of Sciences of the United States of America. 107 (50): 21248–21255. doi:10.1073/pnas.1015855107. PMC 3003076. PMID 21081700.
- ↑ Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA, et al. (June 2015). "MET is required for the recruitment of anti-tumoural neutrophils". Nature. 522 (7556): 349–353. Bibcode:2015Natur.522..349F. doi:10.1038/nature14407. PMC 4594765. PMID 25985180.
- ↑ Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R (September 2011). "Tumor entrained neutrophils inhibit seeding in the premetastatic lung". Cancer Cell. 20 (3): 300–314. doi:10.1016/j.ccr.2011.08.012. PMC 3172582. PMID 21907922.
- ↑ Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. (September 2009). "Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN". Cancer Cell. 16 (3): 183–194. doi:10.1016/j.ccr.2009.06.017. PMC 2754404. PMID 19732719.
- ↑ Coffelt SB, Wellenstein MD, de Visser KE (July 2016). "Neutrophils in cancer: neutral no more" (PDF). Nature Reviews. Cancer. 16 (7): 431–446. doi:10.1038/nrc.2016.52. PMID 27282249. S2CID 4393159.
- ↑ Gabrilovich DI (January 2017). "Myeloid-Derived Suppressor Cells". Cancer Immunology Research. 5 (1): 3–8. doi:10.1158/2326-6066.CIR-16-0297. PMC 5426480. PMID 28052991.
- ↑ Turcotte S, Rosenberg SA (2011). "Immunotherapy for metastatic solid cancers". Advances in Surgery. 45: 341–360. doi:10.1016/j.yasu.2011.04.003. PMC 3578602. PMID 21954698.
- ↑ Clayton A, Tabi Z (May–June 2005). "Exosomes and the MICA-NKG2D system in cancer". Blood Cells, Molecules & Diseases. 34 (3): 206–213. doi:10.1016/j.bcmd.2005.03.003. PMID 15885603.
- ↑ Plitas G, Rudensky AY (March 2020). "Regulatory T Cells in Cancer". Annual Review of Cancer Biology. 4: 459–77. doi:10.1146/annurev-cancerbio-030419-033428.
- ↑ Freedman LP, Gibson MC, Ethier SP, Soule HR, Neve RM, Reid YA (June 2015). "Reproducibility: changing the policies and culture of cell line authentication". Nature Methods. 12 (6): 493–497. doi:10.1038/nmeth.3403. PMID 26020501. S2CID 20557369.
- ↑ Franchi-Mendes T, Eduardo R, Domenici G, Brito C (September 2021). "3D Cancer Models: Depicting Cellular Crosstalk within the Tumour Microenvironment". Cancers. 13 (18): 4610. doi:10.3390/cancers13184610. PMC 8468887. PMID 34572836.
- ↑ Maulana TI, Kromidas E, Wallstabe L, Cipriano M, Alb M, Zaupa C, et al. (June 2021). "Immunocompetent cancer-on-chip models to assess immuno-oncology therapy". Advanced Drug Delivery Reviews. 173: 281–305. doi:10.1016/j.addr.2021.03.015. PMID 33798643.
- 1 2 Zhang J, Tavakoli H, Ma L, Li X, Han L, Li X (August 2022). "Immunotherapy discovery on tumor organoid-on-a-chip platforms that recapitulate the tumor microenvironment". Advanced Drug Delivery Reviews. 187: 114365. doi:10.1016/j.addr.2022.114365. PMID 35667465. S2CID 249381011.
- ↑ Morton JJ, Bird G, Refaeli Y, Jimeno A (November 2016). "Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap". Cancer Research. 76 (21): 6153–6158. doi:10.1158/0008-5472.CAN-16-1260. PMC 5093075. PMID 27587540.
- ↑ Chen A, Neuwirth I, Herndler-Brandstetter D (May 2023). "Modeling the Tumor Microenvironment and Cancer Immunotherapy in Next-Generation Humanized Mice". Cancers. 15 (11): 2989. doi:10.3390/cancers15112989. PMC 10251926. PMID 37296949.
- ↑ Feng Y, Liao Z, Zhang H, Xie X, You F, Liao X, et al. (January 2023). "Emerging nanomedicines strategies focused on tumor microenvironment against cancer recurrence and metastasis". Chemical Engineering Journal. 452: 139506. doi:10.1016/j.cej.2022.139506. S2CID 252676223.
- ↑ Raju GS, Pavitra E, Varaprasad GL, Bandaru SS, Nagaraju GP, Farran B, et al. (June 2022). "Nanoparticles mediated tumor microenvironment modulation: current advances and applications". Journal of Nanobiotechnology. 20 (1): 274. doi:10.1186/s12951-022-01476-9. PMC 9195263. PMID 35701781.
- ↑ Unezaki S, Maruyama K, Hosoda JI, Nagae I, Koyanagi Y, Nakata M, Ishida O, Iwatsuru M, Tsuchiya S (22 November 1996). "Direct measurement of the extravasation of polyethyleneglycol-coated liposomes into solid tumor tissue by in vivo fluorescence microscopy". International Journal of Pharmaceutics. 144 (1): 11–17. doi:10.1016/S0378-5173(96)04674-1.
- ↑ Lilavivat S, Sardar D, Jana S, Thomas GC, Woycechowsky KJ (August 2012). "In vivo encapsulation of nucleic acids using an engineered nonviral protein capsid". Journal of the American Chemical Society. 134 (32): 13152–13155. doi:10.1021/ja302743g. PMID 22827162.
- ↑ Ramishetti S, Huang L (December 2012). "Intelligent design of multifunctional lipid-coated nanoparticle platforms for cancer therapy". Therapeutic Delivery. 3 (12): 1429–1445. doi:10.4155/tde.12.127. PMC 3584330. PMID 23323560.
- ↑ Jain RK (June 1987). "Transport of molecules in the tumor interstitium: a review". Cancer Research. 47 (12): 3039–3051. PMID 3555767.
- ↑ Li C, Jiang P, Wei S, Xu X, Wang J (July 2020). "Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects". Molecular Cancer. 19 (1): 116. doi:10.1186/s12943-020-01234-1. PMC 7367382. PMID 32680511.
- ↑ Maor Y, Yu J, Kuzontkoski PM, Dezube BJ, Zhang X, Groopman JE (July 2012). "Cannabidiol inhibits growth and induces programmed cell death in kaposi sarcoma-associated herpesvirus-infected endothelium". Genes & Cancer. 3 (7–8): 512–520. doi:10.1177/1947601912466556. PMC 3527984. PMID 23264851.
- ↑ Sadelain M, Rivière I, Brentjens R (January 2003). "Targeting tumours with genetically enhanced T lymphocytes". Nature Reviews. Cancer. 3 (1): 35–45. doi:10.1038/nrc971. PMID 12509765.
- ↑ Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS (June 2022). "CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities". Molecular Therapy Oncolytics. 25: 69–77. doi:10.1016/j.omto.2022.03.009. PMC 8980704. PMID 35434273.
- ↑ Globerson-Levin A, Waks T, Eshhar Z (May 2014). "Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells". Molecular Therapy. 22 (5): 1029–1038. doi:10.1038/mt.2014.28. PMC 4015244. PMID 24572294.
- ↑ Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. (September 2015). "Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma". Clinical Cancer Research. 21 (18): 4062–4072. doi:10.1158/1078-0432.CCR-15-0428. PMC 4632968. PMID 26059190.
- ↑ Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. (January 2018). "Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma". Molecular Therapy. 26 (1): 31–44. doi:10.1016/j.ymthe.2017.10.002. PMC 5763077. PMID 29103912.