β-酮硫解酶缺乏症
β-酮硫解酶缺乏症是一种罕见的常染色体隐性代謝疾病,全世界仅报告有50至60例。患者机体无法正确处理異亮氨酸或脂质分解产物[1][2],典型发作年龄为6个月至24个月。
| β-酮硫解酶缺乏症 | |
|---|---|
| 同义词 | 线粒体乙酰乙酰基辅酶A硫解酶缺乏症 |
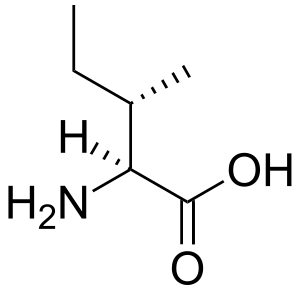 | |
| 异亮氨酸 | |
| 类型 | 常染色体隐性遗传病[*]、congenital disorder of amino acid metabolism[*]、inborn disorder of ketolysis[*]、classic organic aciduria[*]、amino acid metabolic disorder[*] |
| 分类和外部资源 | |
| OMIM | 203750 |
| DiseasesDB | 29824 |
| Orphanet | 134 |
该病症由ACAT1基因突变导致。ACAT1基因产生的酶对分解食物中的蛋白质和脂肪有重要作用。此酶负责加工异亮氨酸,以及脂肪分解过程中产生的酮。ACAT1基因突变会使此酶活性降低或失活,进而导致患者无法正常处理异亮氨酸和酮,有害化合物聚积,血液过于酸性(酮酸中毒),组织功能受到损害,尤以中樞神經系統为甚。
参考资料
This article is issued from Wikipedia. The text is licensed under Creative Commons - Attribution - Sharealike. Additional terms may apply for the media files.