常染色体显性多囊肾
常染色体显性多囊肾(Autosomal dominant polycystic kidney disease,ADPKD)又称为成人型多囊肾,是一种遗传性全身性疾病,主要影响肾脏,但也可能会影响其他器官,如肝脏、胰腺、脑动脉血管等。患有这种疾病的人大约有一半将会发展为终末期肾脏疾病,需要进行透析或肾移植。患者进展为终末期肾病通常发生在40-60岁之间[1]。常染色体显性多囊肾病在全球范围都有发生,发病率大约为1/400人-1/1000人[2][3]。
| 常染色体显性多囊肾 Autosomal dominant polycystic kidney | |
|---|---|
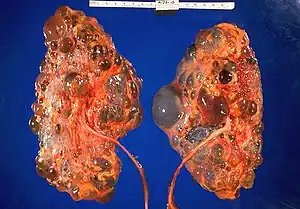 | |
| 多囊肾病理标本 | |
| 类型 | 多囊性腎病變 |
| 分类和外部资源 | |
| 醫學專科 | 醫學遺傳學 |
| ICD-10 | Q61 |
| ICD-9-CM | 753.1 |
| OMIM | 601313 173910 |
| DiseasesDB | 10262 |
| MedlinePlus | 000502 |
| eMedicine | radio/68 |
| MeSH | D016891 |
| Orphanet | 730 |
现在认为,常染色体显性多囊肾和两个基因缺陷有关。85%的患者是由位于16号染色体的基因PKD1(TRPP1)发生突变所致,15%的患者是由4号染色体的PKD2(TRPP2)突变所致[1]。
常染色体显性多囊肾需要和常染色体隐性多囊肾进行辨别。常染色体隐性多囊肾也导致肾脏和肝脏的囊肿,但通常只发生在童年,发病率约为1/20000人[4],病因和预后都和常染色体显性多囊不同。
病理生理学
近期应用实验模式生物如线虫(秀丽隐杆线虫)和家鼠的纤毛和鞭毛进行基本细胞生物学研究使人类发生常染色体显性多囊肾的原因变得清楚。生化学家James Calvet写道:“这些发现证明了遗传学和动物模型的能力和重要性。如果没有这些遗传学研究的指引,有谁会想到一条小小的纤毛会成为研究多囊肾病的基础?”[5]
纤毛在肾脏发育中发挥重要作用,纤毛结构功能异常直接导致肾囊肿性疾病的发生。所有的纤毛和鞭毛的装配和维持都需要依赖鞭毛内运输这个非常重要的生理过程来完成。鞭毛内运输是蛋白质插入纤毛和鞭毛膜特定位点必不可少的一项细胞功能。这些插入的膜蛋白可以启动环境反馈和细胞内信号转导通道。它们在肾小管上皮细胞的纤毛中发挥特殊作用,通过上述鞭毛内运输机制定位于肾小管上皮细胞的纤毛,被认为是正常肾细胞的发育和发挥功能的关键。纤毛上皮细胞排列于尿收集管的内腔,感觉尿流率的变化。纤毛感受尿流率的功能异常可引起肾小管上皮细胞的程序性细胞死亡(凋亡),产生常染色体显性多囊肾特征性的多囊肿。常染色体显性多囊肾可能由于环境信号的转导蛋白或感受蛋白发生突变所致,也有可能是鞭毛内运输失败的结果[6]。
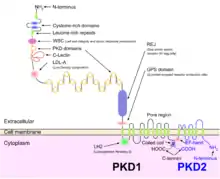
常染色体显性多囊肾基因——PKD1和PKD2编码肾管道细胞非运动纤毛上的膜蛋白。其中PKD2编码的多囊蛋白2(polycystin-2,PC-2)是一个钙激活型细胞内钙释放通道,可以使细胞外的钙离子进入细胞。而PKD1编码的多囊蛋白1(polycystin-1,PC-1)被认为是和PC-2有关,可以调节PC-2钙通道的活性。钙离子是一个重要的细胞信号,可以引发复杂的生化途径,导致细胞休眠和分化。PC-1或者PC-2失去活性,在肾管道细胞纤毛上的装配或者定位失败,对纤毛细胞钙信号的监管放松,都可能导致常染色体显性多囊肾的发生。
常染色体显性多囊肾患者虽然出生时就已经发生基因突变,但却在以后的生活中才出现多囊肾病的表现。这可以用二次打击假说来解释。二次打击假说又称为Knudson假说,是在癌症研究中借来的,在肿瘤发生前致癌基因的两个拷贝都处于休眠状态。在常染色体显性多囊肾中,第一次“打击”是先天性的(无论是PKD1或者是PKD2出现突变),第二次“打击”是指在以后的生活中细胞的生长和分裂。常染色体显性多囊肾的二次打击假说在1992年由Reeders首先提出,并已经被国外多项研究所证实。如常染色体隐性多囊肾患者在出生时就发病,而囊肿衬里上皮细胞正常的PKD1或PKD2基因拷贝中可以发现体细胞突变。
诊断
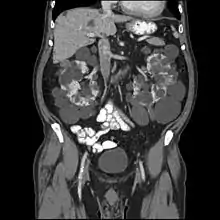
常染色体显性多囊肾的诊断主要依靠影像学或分子遗传学检测来明确。在年龄30岁以上的患者或PKD1突变的年轻患者,检测的敏感度接近100%;而在30岁以下的PKD2突变年轻患者,检查的敏感度仅有67%。婴儿或者儿童在影像学检查中没有看到明显的囊肿,仅有高回声肾的表现发生常染色体显性多囊肾的危险为50%。在没有常染色体显性多囊肾家族史的人,出现双肾肿大和囊肿,伴有或没有伴有肝囊肿,并且不存在其他表现的,提示可能肾囊肿性疾病,但并非诊断明确的证据。
连锁分析或直接突变筛查等分子遗传检测有临床价值。但是遗传异质性是分子遗传检测的一个障碍。有时候要检测一个家族中的很多成员才可以确定哪一个基因才是这个家族致病的原因。PKD1和PKD2基因的较大,并且非常复杂,具有明显的等位基因异质性,这些都是目前DNA分子检测的障碍。在研究环境中,PKD1基因的突变检测率为50-75%,而PKD2基因突变检测率为-75%。现在已经有用于临床的PKD1和PKD2的直接序列分析,可以检测出50-70%的致病突变。
遗传咨询可能有助于有得多囊肾病风险的家庭。
治疗
tolvaptan是目前唯一经过批准用于延缓囊肿扩大的药物。
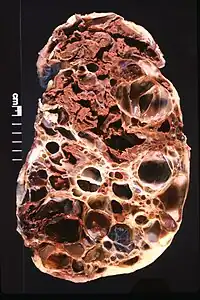
虽然常染色体显性多囊肾是无法治愈的,但对症治疗可以缓解症状,延长患者的生命。
- 疼痛:非处方止痛药如扑热息痛得不发,可以减轻患者的疼痛。对于大多数(但并非所有)患者剧烈疼痛的症状,可以通过手术缩小囊肿以减轻背部和侧面的疼痛。然而,手术只能临时缓解症状,通常不延缓肾功能衰竭的进展。
- 泌尿道感染:常染色体显性多囊肾患者往往有反复的尿路感染症状,可以用抗生素治疗。尿路感染最重要的要求是早期治疗,因为尿路感染可以沿尿道蔓延至肾囊肿,而囊肿感染是很难治疗的,因为许多抗生素无法渗透到囊肿中。不过仍然有部分抗生素是有效的。
- 高血压:控制血压可以缓慢常染色体显性多囊肾的影响。生活方式的变化,包括低盐饮食和使用各种药物,尤其是血管紧张素转化酶抑制剂和血管紧张素II受体拮抗剂等可以降低患者的高血压,推荐的目标血压是降到80/130 mmHg或更低。
- 终末期肾病:有两种选择恢复患者的肾功能:透析或肾移植。健康(非常染色体显性多囊肾)的肾脏移植到患者体内后不会出现囊肿。
预后
尽管对常染色体显性多囊肾研究进展很快,但本病的预后变化从发现至今不大。有人认为避免摄入咖啡因可以防止形成囊肿。治疗高血压和低蛋白饮食可减缓疾病的进展,但没有得到很好的验证。
PKD1基因突变导致的常染色体显性多囊肾预后比PKD2基因突变的更差。
参考资料
- Grantham, Jared. . New England Journal of Medicine. Oct 2, 2008, 359: 1477–1485 [2011-09-27]. doi:10.1056/NEJMcp0804458. (原始内容存档于2022-06-15).
- Torres, Vicente. . Kidney International. 20 May 2009, 76 (2): 149–168 [2011-09-27]. doi:10.1038/ki.2009.128. (原始内容存档于2015-09-23).
- DALGAARD OZ. . Acta Med. Scand. Suppl. 1957, 328: 1–255. PMID 13469269.
- Zerres K, Mücher G, Becker J; et al. . Am. J. Med. Genet. 1998, 76 (2): 137–44. PMID 9511976. doi:10.1002/(SICI)1096-8628(19980305)76:2<137::AID-AJMG6>3.0.CO;2-Q.
- Calvet, James P. . Journal of the American Society of Nephrology. Oct 2002, 13 (10): 2614–16 [2011-09-27]. PMID 12239253. (原始内容存档于2007-02-13).
- Yoder BK, Hou X, Guay-Woodford LM. . Journal of the American Society of Nephrology. Oct 2002, 13 (10): 2508–2516. PMID 12239239. doi:10.1097/01.ASN.0000029587.47950.25.
{kind=link}
{kind=link}