手性助剂
手性助劑是一種為了控制立體化學的合成結果而暫時加入到有機合成反應中的化合物或單元。[1][2]手性作為一個輔助劑可以偏置一或更多後續反應(subsequent reactions)的立體選擇性。通常輔助劑可以從基板(substrate)上切下然後回收以供將來使用。

大部分的生物分子和標靶藥物存在著兩種可能的鏡像異構物(又稱對映異構)中的其中一種;因此,天然產物和藥物試劑的化學合成通常被設計成獲得目標在單一鏡像化合物(enantiomerically pure)的形式。[3]化學家會使用很多方法去合成所期望的立體異構物,而加入手性助劑即為方法之一。[4]
1975年艾里亞斯·詹姆斯·科里用手性8-苯基薄荷醇(chiral 8-phenylmenthol)來介紹何謂手性助劑,而後在1980年巴里·特羅斯特也以手性扁桃(chiral mandelic acid)酸來介紹之。但因薄荷醇的製備困難,所以在1985年J. K. Whitesell以反-2-苯基-1-環己醇(trans-2-phenyl-1-cyclohexanol)將其取代。
非對稱合成
為了控制化合物立體中心的絕對構造,手性助劑會在合成途徑中被結合。在眾多使用手性助劑的合成中大衛·A·伊凡斯cytovaricin的合成被認為是經典。他利用一個不對稱烷基化反應和四個不對稱羥醛縮合反應而成的噁唑烷酮手性輔助劑,產生出有9個立體中心的絕對立體化學結構。[5]

一個典型反應在輔助引導下的立體選擇性轉化需要三個步驟:首先,由輔助劑共價結合在基板上;接著,所產生的化合物會進行一或多種非鏡像選擇性(diastereoselective)的轉換;而最後,輔助劑在不會引起所需產物外消旋化(racemization)的情況下被移除。[4]使用化學劑量輔助劑花費的能量加上合成步驟中結合和除去所需的能量使得此途徑似乎較沒效效率。然而,對許多轉化物來說,唯一可以產生立體選擇性的方法必須依賴手性助劑。此外,使用手性助劑的轉換趨於多用途且好研究,從而使得單一鏡像異構物的獲得變得最為省時。[2]
更進一步說明,[6]在輔助劑引導反應下的產物為非鏡像異構物(或稱非對映異構),能使他被一些方法如管柱層析法(column chromatography)或是結晶做簡單分離。
8-苯基薄荷醇
在先前手性助劑使用於不對稱合成的範例中,艾里亞斯·詹姆斯·科里與同事進行(-)-8-苯基薄荷醇丙烯酸酯和5-苄氧甲基環戊二烯(5-benzyloxymethylcyclopentadiene)之間的不對稱雙烯加成反應(狄耳士–阿德爾反應)。[7]而環化反應後的產物碘代內酯(iodolactone)如下圖表示,為典型Corey反應中,前列腺素合成的中間產物。因為丙烯酸酯的背面被輔助劑阻擋,所以環化反應只能發生在烯烴的前方。
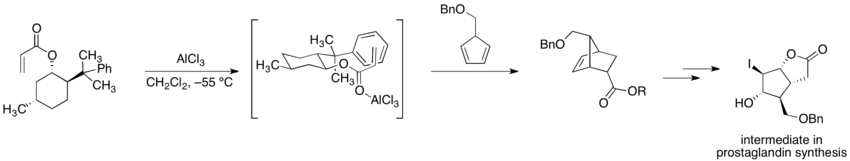
(-)-8-苯基薄荷醇可以從任一長葉薄荷酮的鏡像異構物中製備,[8]雖然沒有一個途徑是比較有效率的。因為8-苯基薄荷醇的廣泛使用,更容易合成的替代性化合物,例如「反」-2-苯基-1-環己醇[9]和反-2-( 1-苯基-1-甲基乙基)環己醇(trans-2-( 1-pheyl-1-methylethyl)cyclohexanol)[10]已經被探究。
噁唑烷酮
噁唑烷酮輔助劑,由大衛·A·伊凡斯普及於世,已經被應用到許多立體選擇性轉換上,包括羥醛縮合反應[11]、烷基化反應,[12]和狄爾斯 - 阿爾德反應(又稱雙烯加成反應)。[13][14]噁唑烷酮取代4和5位置。透過空間位阻(立體阻礙),因此取代基引導了各種基團取代的方向。而輔助劑便隨後被除去,例如:透過水解的方式。
製備
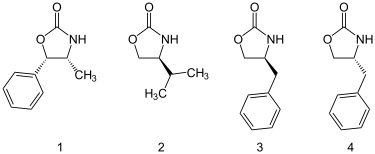
噁唑烷酮可以從胺基酸或從容易取得的氨基醇中製備出。大量的噁唑烷酮是有市售的,包括以下四種。

烷基化反應
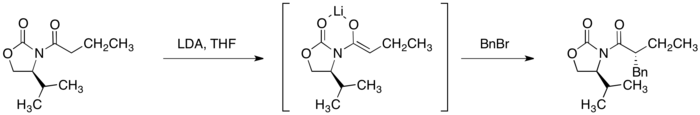
噁唑烷酮酰亞胺加上強鹼,像是二異丙基氨基鋰能選擇性提供(Z)-烯醇的強鹼,噁唑烷酮酰亞胺的α-碳去質子化,最終它可以進行立體選擇性烷基化。
羥醛縮合
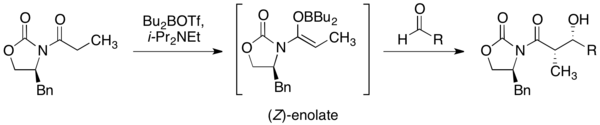
噁唑烷酮輔助劑在立體選擇性的羥醛縮合反應是最為廣泛使用的。使用三氟甲磺酸二丁硼作為路易士酸和二異丙基乙胺作為鹼所形成的軟烯醇化反應,會產生出(Z)-烯醇化物((Z)-enolate)。而此化合物可用醛當作基板形成非鏡像選擇性的羥醛縮合反應。這種轉化是非常有能量的,因為它形成了兩個連續的立體結構中心。
觀察下面的範例圖形,甲基團和新二級醇的”同相位”立體關係是因為Zimmerman-Traxler六元環過渡態。醛的取向使得氫被放置在偽軸向的取向上,為的就是要避免1,3-雙軸性交互作用。而雙立體結構中心的絕對立體結構是取決於輔助劑的手性。在過渡結構中,輔助劑羥基會被導向遠離烯醇氧,以盡量減少分子間的淨偶極;手性助劑上的取代基就會阻擋住烯醇化合物的其中一面。

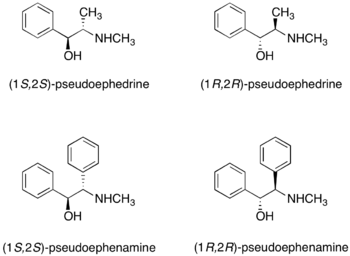
硫酸偽麻黃鹼
(R,R)-和(S,S)-硫酸偽麻黃鹼皆可作為手性助劑。[15]硫酸偽麻黃鹼與羧酸、酸酐和醯氯反應形成硫酸偽麻黃醯胺(pseudoephedrine amide)。
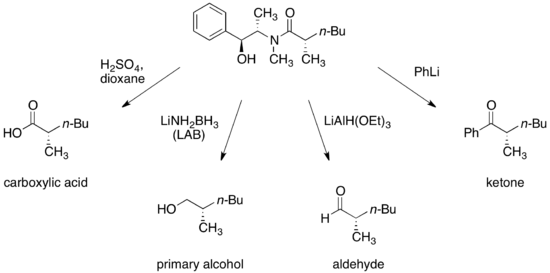
羰基的α-質子很容易被非親核鹼移除,而近一步形成烯醇化物(enolate)。加成化合物的構造是被甲基所導向的,例如鹵代烷。因此,任何加成產物與甲基成反相位,而與羥基成同相位。硫酸偽麻黃鹼手性助劑的後續移除,是藉由適當的親核試劑切除醯胺鍵而完成。
製備
硫酸偽麻黃鹼的兩種鏡像異構物皆可市售。外消旋硫酸偽麻黃鹼是市售的蘇達菲以及其他品牌的鼻腔解充血劑。因為硫酸偽麻黃鹼可以轉化為非法物質甲基苯丙胺,所以不管是學術用途或工業研究上的購買,都是受到監管的。作為替代,Myers和同事最近發表了pseudoephenamine手性助劑在烷基化反應中的效用。[16]然而pseudoephenamine不容易從商業資源中取得,他可以從其他可獲得的物質中合成,而不是像硫酸偽麻黃鹼一樣被管制。

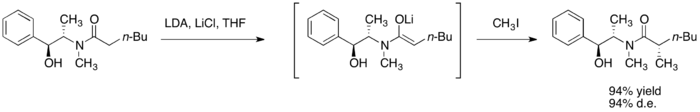
烷基化
硫酸偽麻黃醯胺藉由強鹼如二異丙基胺基鋰(LDA),進行去質子反應後,而得到相應的(Z)-烯醇。這些烯醇鋰的烷基化擁有高面相選擇性的回收率。
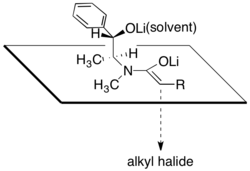
硫酸偽麻黃醯胺的非鏡像選擇性被認為是結構所造成的,因為烯醇鋰的其中一個面向,會被醇化鋰和與鋰陽離子有關的溶劑分子阻擋住。根據此說法指出,在烷基化步驟的非鏡像選擇性高度依賴於氯化鋰的存在量與所使用的溶劑,四氫呋喃(THF)。一般來說,4到6當量的氯化鋰就足以讓烯醇化物在THF溶液中的反應莫耳濃度達到飽和。
使用硫酸偽麻黃醯胺進行烷基化的一個主要優點,是醯胺烯醇的親和性通常足夠與一級、甚至二級鹵化物發生反應,而此反應在溫度-78 °C到0 °C 下進行。也可藉由醯胺烯醇的α-支鏈烷基化將四元碳結構放置中央,但因親電子試劑的反應性較低,所以必須加入DMPU (页面存档备份,存于)。[18]
三-丁基亞磺醯胺
Jonathan Ellman已經廣泛研究出利用手性亞磺醯胺(sulfonamide)的衍生物作為手性助劑的方法。[19]
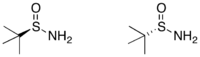
製備
任一「三」-丁基亞磺醯胺的鏡像異構物可藉由兩步驟從「三」-丁基二硫醚(tert-butyl disulfide)得到:以不對稱催化氧化反應得到高產率的二硫化單氧化物和過剩的鏡像異構物。接著以在氨水中的氨化鋰處鋰該化合物,即可得到單一光學性的反向產物。
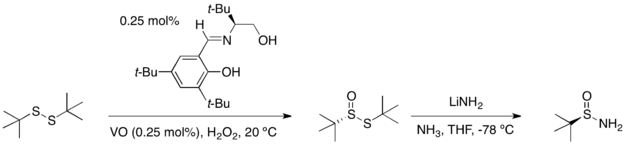
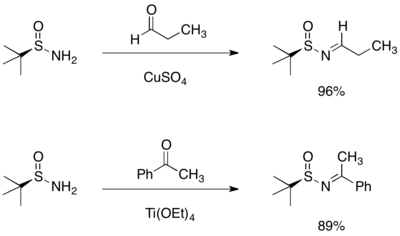
以醛或酮來進行三-丁基亞磺醯胺的縮合反應,會得到高產率的且只有(E)-異構物的相對應醛亞胺和酮亞胺或N-亞磺醯胺。
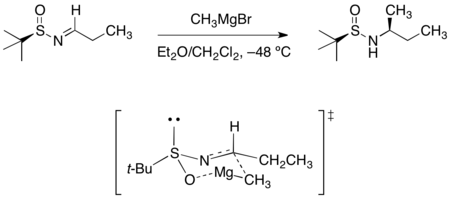
手性胺的合成
在三-丁基亞磺醯醛亞胺(tert-butanesulfinyl aldimine)或氯胺酮(ketamine)中添加Grignard試劑會導致不對稱加成,而得到之鏈亞磺醯胺。所觀察到的立體選擇性可被六元環的過渡結構合理化,其中亞磺醯亞胺的氧與氮皆會與鎂做結合。
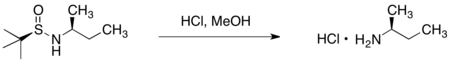
SAMP/RAMP
手性(S)-1-胺基-2-甲氧基甲基吡咯啶(SAMP)和(R)-1-胺基-2-甲氧基甲基吡咯啶(RAMP)的腙烷基化反應,是由迪特·恩德斯和艾里亞斯·詹姆斯·科里研發。[20][21]
製備
SAMP可藉由(S)-脯胺酸((S)-proline)以六個步驟製備,而RAMP則可藉由(R)-谷胺酸((R)-glutamic acid)以六個步驟製備。
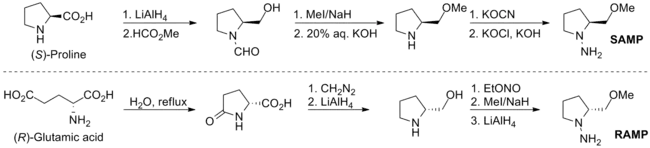
烷基化反應
以醛或酮進行SAMP或RAMP的縮合反應會得到(E)-聯氨類,而以鋰二異丙基醯胺(lithium diisopropylamide)去質子化以及鹵化烷加成,就會得到烷基化合物。此反應的輔助劑可由臭氧化反應或水解。
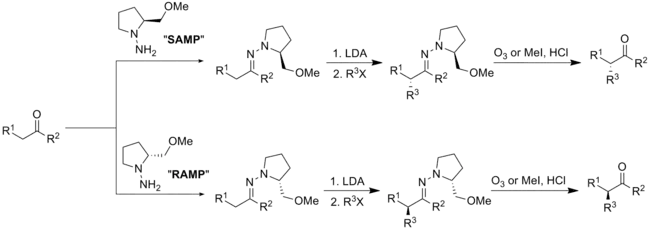
工業上的手性助劑
手性助劑通常是可靠且多功能的,使得大量的單一鏡像異構物可在省時的方式下合成。因此,手性助劑經常會在藥物開發的初期階段被使用。[2]

另請參見
- 反式-2-苯基-1-環己醇(trans-2-Phenyl-1-cyclohexanol)作為手性助劑的使用範例:尾島內醯胺
- 纈胺酸在Schöllkopf方法中擔任手性助劑。
參考
- Key Chiral Auxiliary Applications (Second Edition)(ed.: Roos, G.), Academic Press, Boston, 2014. ISBN 978-0-12-417034-6.
- Glorius, F.; Gnas, Y. . Synthesis. 2006, 12: 1899–1930. doi:10.1055/s-2006-942399.
- Jamali, Fakhreddin. . Wainer, Irving W. (编). . Marcel Dekker, Inc. 1993: 375–382. ISBN 0-8247-8819-2.
- Evans, D. A.; Helmchen, G.; Rüping, M. . Christmann, M (编). . Wiley-VCH Verlag GmbH & Co. 2007: 3–9. ISBN 978-3-527-31399-0.
- Nicolau, K. C. 5th. New York, New York: Wiley-VCH. 2008: 485–508. ISBN 978-3-527-29231-8.
- Miller, J. P. . ChemInform. 2013, 44 (48). doi:10.1002/chin.201348243.
- Corey, E. J.; Ensley, H. E. . J. Am. Chem. Soc. 1975, 97 (23): 6908–6909. doi:10.1021/ja00856a074.
- Corey, E. J.; Ensley, H. E.; Parnell, C. A. . J. Org. Chem. 1978, 43 (8): 1610–1611. doi:10.1021/jo00402a037.
- Whitesell, J. K.; Chen, H. H.; Lawrence, R. M. . J. Org. Chem. 1985, 50 (23): 4663–4664. doi:10.1021/jo00223a055.
- Comins, D. L; Salvador, J. D. . J. Org. Chem. 1993, 58 (17): 4656–4661. doi:10.1021/jo00069a031.
- Evans, D. A.; Bartroli, J.; Shih, T. L. . J. Am. Chem. Soc. 1981, 103 (8): 2127–2129. doi:10.1021/ja00398a058.
- Evans, D. A.; Ennis, M D.; Mathre, D. J. . J. Am. Chem. Soc. 1982, 104 (7): 1737–1739. doi:10.1021/ja00370a050.
- Evans, D. A.; Chapman, K. T.; Bisaha, J. . J. Am. Chem. Soc. 1984, 106 (15): 4261–4263. doi:10.1021/ja00327a031.
- Evans, D. A.; Chapman, K. T.; Hung, D. T.; Kawaguchi, A. T. . Angew. Chem. Int. Ed. 1987, 26 (11): 1184–1186. doi:10.1002/anie.198711841.
- Myers, A. G., et al., Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones, J. Am. Chem. Soc., 1997, 119, 6496-6511.doi:10.1021/ja970402f
- Myers, A. G.; Morales, M. R.; Mellem, K. T. . Angew. Chem. 2012, 124: 4646–4649. doi:10.1002/ange.201200370.
- Myers, A. G.; Yang, B. H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. . J. Am. Chem. Soc. 1997, 119: 6496–6511. doi:10.1021/ja970402f.
- Kummer, D. A.; Chain, W. J.; Morales, M. R.; Quiroga, O.; Myers, A. G. . J. Am. Chem. Soc. 2008, 130: 13231–13233. doi:10.1021/ja806021y.
- Ellman, J. A.; Owens, T. D.; Tang, T. P. . Acc. Chem. Res. 2002, 35: 984–995. doi:10.1021/ar020066u.
- Corey, E. J., Enders, D. . Tetrahedron Letters. 1976, 17 (1): 3–6. doi:10.1016/s0040-4039(00)71307-4.
- Kurti, L.; Czako, B. . Burlington, MA: Elsevier Academic Press. 2005: 150–151. ISBN 0-12-369483-3.
- Turner, S. T.; et al. . J. Med. Chem. 1998, 41: 3467–3476. doi:10.1021/jm9802158.
- Caron, Stéphane. . Caron, Stéphane (编). . John Wiley & Sons, Inc. 2011: 666–670. ISBN 978-0-470-03733-1.
- Roth, B. D. et al.' '. . J. Med. Chem. 1991, 34: 357–366. doi:10.1021/jm00105a056.
- Jie Jack Li, Douglas S. Johnson, Drago R. Sliskovic, Bruce D. Roth. . . John Wiley & Sons, Inc. 2004: 113–125. ISBN 0-471-21480-9.