晶体场理论
晶体场理论(英語: CFT)是一种配位化学理论。
它以过渡金属配合物的电子层结构为出发点,可以很好地解释配合物的磁性、颜色、立体构型、热力学性质和配合物畸变等主要问题,但不能合理解释配体的光谱化学序列和一些金属有机配合物的形成。
1928年,研究晶体光学的德裔美国物理学家汉斯·贝特[1]提出了晶体场理论,他从静电场出发,揭示了过渡金属元素配合物晶体的一些性质。1932年美国物理学家范·弗莱克[2]发展了这一理论。
晶体场理论认为中心阳离子对阴离子或偶极分子负端的静电吸引类似于离子晶体中的正、负离子之间的相互作用[3],着眼于中心原子的d轨道在各种对称性配位体静电场中的变化,简明直观,方便结合实验数据进行定量或半定量的计算。
因为在实际配合物中纯离子键或纯共价键都比较罕见,现代配合物结构理论兼有晶体场理论和分子轨道理论的优点,形成了配位场理论。
概述
晶体场理论认为,配合物中心原子处在配体所形成的静电场中,两者之间完全靠静电作用结合,类似于正负离子之间的作用。在晶体场影响下,五个简并的d轨道发生能级分裂,d电子重新分布使配合物趋于稳定。
能级分裂
d原子轨道分为、、、和五种,其空间取向各不相同,但能级却是相同的,参见原子轨道。在一定对称性的配位体静电场(负)作用下,由于与配位体的距离不同,d轨道中的电子将不同程度地排斥配位体的负电荷,d轨道开始失去简并性而发生能级分裂。能级分裂与以下因素有关:





- 金属离子的性质;
- 金属的氧化态,高氧化态的分裂能较大;
- 配合物立体构型,即配体在金属离子周围的分布;
- 配位体的性质。
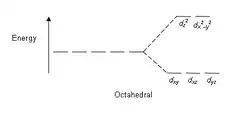
最常见的配合物构型为八面体,其中中心原子位于八面体中心,而六个配位体则沿着三个坐标轴的正、负方向接近中心原子。
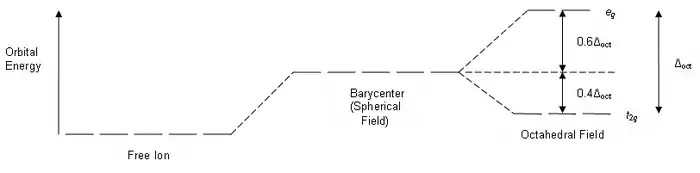
先将球形场的能级记为。和轨道的电子雲极大值方向正好与配位体负电荷迎头相碰,排斥较大,因此能级升高较多,高于。而、和轨道的电子雲则正好处在配位体之间,排斥较小,因此能级升高较小,低于。

因而d轨道分裂为两组能级:
- 和轨道,能量高于,记为或轨道;
- 、和轨道,能量低于,记为或轨道。




和是来自于群论的对称性符号。两组轨道之间的能级差记为Δ0或10,称为分裂能。

量子力学指出,虽然晶体场对称性可能有变化,但受到微扰的d轨道的平均能量是不变的,等于能级。选取能级为计算零点,则有:
- - = 10
- 2 + 3 = 0


解得:
- = 6
- = -4
也就是说,正八面体场中d轨道能级分裂的结果是,与能级相比,轨道能量升高6(0.6Δ0),而轨道能量则降低了4(0.4Δ0)。
类似地可求出正四面体场中的能级分裂结果:
- 由、和轨道组成,高于1.78;
- 由和轨道组成,低于2.67。


平面正方形场(假设为xy平面):
- 轨道——12.28
- 轨道——2.28
- 轨道——-4.28
- 和轨道——-5.14
其他构型的分裂情况请参考#能级分裂图。
分裂能
分裂能Δ的大小既与配体有关,也与中心原子有关。
- 中心原子一定时,Δ随配体不同而改变,主要遵循以下的顺序:
- I− < Br− < S2− < SCN− < Cl− < NO3− < N3− < F− < OH− < C2O42− < H2O < NCS− < CH3CN < py < NH3 < en < 2,2'-bipy < phen < NO2− < PPh3 < CN− < CO
- 由于Δ值由光谱确定,故该顺序也称为光谱化学序列,用以表示配体场强度顺序。该顺序可用配位场理论来解释。
- 配体一定时,Δ随中心原子改变。
- 同一元素中心原子电荷越大时,Δ值也越大。
- 不同周期的中心原子,Δ值随周期数增大而增大。
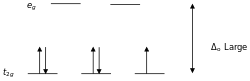
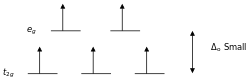
- Δ值随配位原子原子半径减小而增大:I < Br < Cl < S < F < O < N < C。
高自旋和低自旋
当某个d轨道中已经有一个电子时,若第二个电子继续进去与其配对,则电子成对能P可定义为第二个电子克服第一个电子排斥作用所需的能量。在d轨道能级分裂后,d电子的排布要兼顾能量最低原理和洪德规则,既要尽可能分占不同轨道且自旋平行,还要保证总体能量最低,因而最终取决于分裂能Δ和电子成对能P的相对大小。
根据光谱化学序列,在CN−和CO等强场配体作用下,配合物分裂能更大,Δ>P,d电子更易在能级低的轨道中排布,称为低自旋。比如,在强场配体NO2−的作用下,八面体构型离子[Fe(NO2)6]3−的5个d电子将全部处于轨道中。
与此相反,I−与Br−之类的弱场配体导致Δ<P,d电子更易排布在能级高的轨道中,称为高自旋。例如,在含有弱场配体Br−的离子[FeBr6]3−中,5个d电子中有3个处于轨道,2个处于轨道,形成5个单占轨道且为高自旋态。
| d电子数 | 弱场高自旋 | 未成对电子数 | 强场低自旋 | 未成对电子数 | 强场中降低的轨道能量 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ↑ | ↑ | ||||||||||||
| ↑ | ↑ | ↑ | ↑ | ||||||||||
| ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||||
| ↑ | ↑ | ↑ | ↑ | ↑↓ | ↑ | ↑ | |||||||
| ↑ | ↑ | ↑ | ↑ | ↑ | ↑↓ | ↑↓ | ↑ | ||||||
| ↑↓ | ↑ | ↑ | ↑ | ↑ | ↑↓ | ↑↓ | ↑↓ | ||||||
| ↑↓ | ↑↓ | ↑ | ↑ | ↑ | ↑↓ | ↑↓ | ↑↓ | ↑ | |||||
| ↑↓ | ↑↓ | ↑↓ | ↑ | ↑ | ↑↓ | ↑↓ | ↑↓ | ↑ | ↑ | ||||
| ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑ | ||||
| ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ||||
可见,只有、、和的电子组态才有可能有两种不同的排布。




对正四面體型配合物而言,Δ四面體大约只等于Δ八面体,且成对能P变化不大,因此四面体型配合物都符合洪德规则,为高自旋。

“弱场高自旋,强场低自旋”的结论已得到配合物磁性实验的证实,可用于预测未知配合物的磁性性质。含有未成对电子的配合物呈顺磁性,而不含未成对电子的配合物则呈抗磁性。
晶体场稳定化能
假想配体以“球形场”作用于中心原子d轨道不分裂时配合物的能量为0,晶体场稳定化能(CFSE)便是配位场引起d轨道分裂导致配合物能量的下降值。晶体场稳定化能可通过计算各电子所处能级能量的代数和得到,轨道中的电子会使稳定化能减小,而轨道中的电子则会使稳定化能增加。例如,对于八面体场中的组态而言,一个电子从球形场进入轨道,能量下降即晶体场稳定化能为:-4(计算方法参见上文#能级分裂)。

| 电子构型 | 稳定化能 | 电子构型 | 稳定化能 | ||
|---|---|---|---|---|---|
| -4 | 高自旋 | -4 | |||
| -8 | 低自旋 | -24 + 2P | |||
| -12 | 高自旋 | -8 | |||
| 高自旋 | -6 | 低自旋 | -18 + P | ||
| 低自旋 | -16 + P | -12 | |||
| 高自旋 | 0 | -6 | |||
| 低自旋 | -20 + 2P | 0 | |||





晶体场稳定化能也可以解释其他构型的过渡金属配合物,并且很多构型原子的平面正方结构便可归咎于该构型相对较高的稳定化能。需要注意的是,晶体场稳定化能并不是配合物绝对稳定性的量度,并且为零的稳定化能只说明考虑d轨道分裂与不考虑d轨道分裂引起的配体与中心原子d轨道之间的排斥力没有区别,并不表明配合物不具稳定性。
对配合物颜色的解释
晶体场理论还可解释很多配位化合物的颜色。基态的d电子处在较低能量的轨道中,受光照射获得能量时,就会吸收光子跃迁到能量较高的一组,发生d-d跃迁。其显色的基本原因是某些光被吸收而呈现被吸收光的补色,因此,可通过计算吸收光子的能量来进一步预测配合物的颜色。这也是晶体场理论被推崇备至的重要原因之一。
由于不同的配体导致的分裂能大不相同,因此配合物的颜色也有很大差异。金属原子不变时,弱场致使分裂能减小,光的波长增加,频率减小;强场则使分裂能增大,光频率增大,补色频率减小。虽然这只是分裂能不同及配合物显色的原因之一,其他的还包括电荷迁移、电子排斥及姜-泰勒效应等等,但由强场弱场导致的显色差异仍在一些化合物中有所体现。
颜色理论

物体的颜色是被吸收的光的补色。
| λ吸收 | 吸收光 | 呈现颜色 | λ呈现 |
|---|---|---|---|
| 400 nm | 紫色 | 黄绿色 | 560 nm |
| 450 nm | 蓝色 | 橙色 | 600 nm |
| 490 nm | 蓝绿色 | 红色 | 620 nm |
| 570 nm | 黄绿色 | 紫色 | 410 nm |
| 580 nm | 黄色 | 深蓝色 | 430 nm |
| 600 nm | 橙色 | 蓝色 | 450 nm |
| 650 nm | 红色 | 绿色 | 520 nm |
姜-泰勒效应
为了解释某些过渡金属配合物中所出现的拉长或扁平八面体稳定结构,1937年,姜(Jahn H.A)与泰勒(Teller E.)用群论证明:
该现象通常出现在组态的Cu2+中,它含有能量相等的两种排布方式:


因而会发生构型畸变。此外,若畸变发生是因高能级d轨道上的简并态,则变形较大,称为大畸变;反之则称为小畸变。
平面正方形配合物也可看作八面体被拉长的极限情况。的Ni2+、Pt2+易生成平面正方形低自旋配合物,可用采取电子结构,z方向排斥比xy平面方向大得多来解释。

四面体型配合物也可能产生姜-泰勒效应,只是d轨道能级分裂较小,相应的畸变也很小。
能级分裂图
- 注意:下图仅为示意图,图中距离和能级的实际分布不完全一致。
| 八面体 | 五角双锥 | 三角双锥 |
|---|---|---|
 |  | 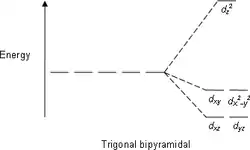 |
| 平面正方 | 四方锥 | 四面体 |
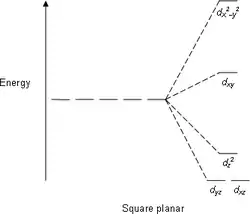 | 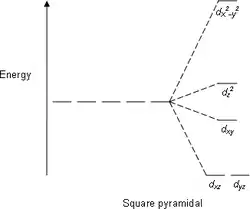 | 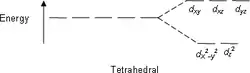 |
各种构型
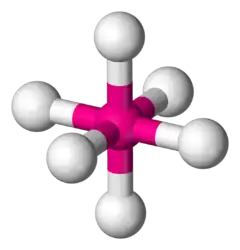
八面体 
五角双锥 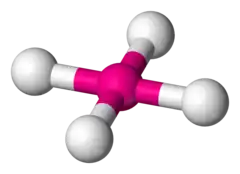
平面正方 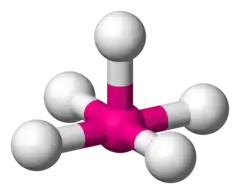
四方锥 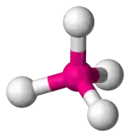
四面体 
三角双锥 
五角锥
参考资料
- Zumdahl, Steven S. Chemical Principles, Fifth Edition. Boston: Houghton Mifflin Company, 2005. 550-551,957-964.
- Silberberg, Martin S. Chemistry: The Molecular Nature of Matter and Change, Fourth Edition. New York: McGraw Hill Company, 2006. 1028-1034.
- D. F. Shriver, P. W. Atkins. Inorganic Chemistry 3rd edition, Oxford University Press, 2001. Pages: 227-236.
- Housecroft, C. E. and Sharpe, A. G. (2005) Inorganic Chemistry 2nd edition, England: Pearson Education Limited. ISBN 0-13-039913-2
- Miessler, G. L. and Tarr, D. A. (2003) Inorganic Chemistry 3rd edition, New Jersey: Pearson Prentice Hall. ISBN 0-13-035471-6
- 张季爽,申成主编。《基础结构化学》第二版。北京:科学出版社,2006年。ISBN 7-03-016425-2
- 北京师范大学无机化学教研室等编。《无机化学》上册。北京:高等教育出版社,2002年。ISBN 7-04-010768-6
- Bethe, H. . Annalen der Physik. 1929, 395 (2): 133–208. Bibcode:1929AnP...395..133B. ISSN 1521-3889. doi:10.1002/andp.19293950202 (德语).
- Van Vleck, J. . Physical Review. 1932, 41 (2): 208–215. Bibcode:1932PhRv...41..208V. doi:10.1103/PhysRev.41.208.
- 张祖德. . 2008: 303–304. ISBN 978-7-312-03560-9.
- G. L. Miessler and D. A. Tarr “Inorganic Chemistry” 2nd Ed. (Prentice Hall 1999), p.379 ISBN 0-13-841891-8.