朱莉娅-科隆纳环氧化反应
朱莉娅-科隆纳环氧化反应()是一个以聚亮氨酸为催化剂的碳-碳双键不对称环氧化反应。该反应在1980年由塞巴斯蒂安·朱莉婭(Sebastian Juliá)报道,[1]以及由朱莉婭和斯蒂法諾·科隆納(Stefano Colonna)同时做的进一步阐述。[2]
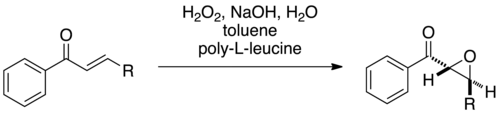
原始的反应模式为三相反应,包含了水相、有机相和催化剂相。作为反应物的查耳酮衍生物溶于有机溶剂(例如甲苯或四氯化碳)中,成为有机相。碱性过氧化氢溶液为水相。反应则发生在既不溶于水相,也不溶于有机相的催化剂聚亮氨酸的表面。两相甚至均相的改进方法也已开发出来,改进后的反应活性和速率均有显著提高。[3][4]
该反应是在温和条件下,立体选择性地环氧化双键的有效手段,因而在有机合成上具有重大的价值:环氧化物不仅是许多有机合成的中间体,许多天然产物也都是过氧化物。此外,经 拜耳 (页面存档备份,存于)(Bayer)和埃佛歷克 (页面存档备份,存于)(Evonik)等人的工作,该反应已经能够有效地应用于工业生产。最后,该反应的催化剂聚氨基酸具有类似酶的性质,因而该反应也有助于对生源合成的研究。[5][6]
反应机理
朱莉娅-科隆纳氧化反应本质上是缺电子碳-碳双键(例如α,β不饱和酮中的碳-碳双键)的不对称亲核环氧化反应。图 2展示了亲核环氧化反应的一般机理,在此反应中,聚亮氨酸作为催化剂控制反应。

过氧化氢阴离子与查耳酮在聚亮氨酸催化剂中形成复合物,然后反应生成过氧化物阴离子中间体。中间体在催化剂结构的控制下迅速关环,立体选择性地形成产物环氧化物。
三元复合物的生成

对该反应的化学动力学研究表明,该反应对含碳-碳双键的底物和过氧化氢负离子都呈现类似酶促反应的动力学,即在底物(或过氧化氢离子)浓度较低时,反应速率与底物(或过氧化氢离子)浓度成正比,而在底物(或过氧化氢离子)浓度较高时,反应速率与浓度无关。两者所对应的米氏常数分别为Km1=110mM和Km2=30mM。 该研究表明,反应经历了一个随机的三元复合物(聚亮氨酸,过氧化氢负离子,底物)的形成过程,并达到稳态。 在形成三元复合物之前,底物和过氧化氢负离子都必须先结合到聚亮氨酸上,虽然两者结合的先后次序可以颠倒,但在动力学上,过氧化氢负离子先结合的过程更为有利。之后三元复合物继续反应生成过氧化物阴离子中间体,这是整个反应的决速步。 (图 3)[5][8]
反应的立体化学
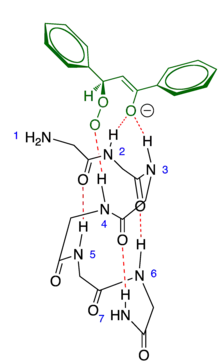
在形成过氧化物阴离子中间体之前,所有的反应物必须与催化剂聚亮氨酸结合。催化剂采取α-螺旋构象。反应中,催化剂分子上的四个位于N端附近的酰胺氢与底物形成氢键,从而固定了底物乃至中间体的空间取向。虽然也有人提出了其他的模型[9]但凱莉(Kelly)等人的计算表明,NH-2, NH-3 和NH-4 形成了等边三角形,可以与中间体(过氧化物阴离子)形成氢键,从而稳定之。(这一行为类似于酶中的氧阴离子穴。) 虽然含碳碳双键的底物既可以从α-螺旋外侧与催化剂结合,也可以从里侧与之结合,只有后一种取向才能将NH-4指向过氧化氢负离子,使得最后一步消除反应成为可能。(图 4)因而,反应是立体专一的。[7]
催化剂

应用范围
这一缺电子碳-碳双键的环氧化反应最初用于查耳酮的氧化。该反应很快即被运用于其他连有吸电子基的碳-碳双键的环氧化反应,例如α,β-不饱和酮,酯,酰胺。[1][2]反应对不饱和的砜也是有效的。[12]
但有些底物则不适宜用该反应环氧化,包括含有会被过氧化氢破坏的基团的物质,α位含有可解离氢的物质,富电子的碳-碳双键。[10]
这一亲核环氧化反应是对亲电环氧化反应(例如Sharpless不对称环氧化反应)的自然补充。
立体选择性
催化剂的结构
反应的立体选择性取决于聚亮氨酸催化剂的α-螺旋结构。如上文所述,虽然N端区域的手性一致是必要的,10个手性一致的亮氨酸构成的多肽已经可以使反应有相当好的立体选择性了。[10]
改进方法
两相 (无水) 反应条件
在两相反应的模式下,底物,作氧化剂的尿素-过氧化氢复合物,作碱的三级胺(例如DBU)溶解在四氢呋喃中,形成一相;固定为胶状物的聚合物催化剂为另一相。这一改进极大地拓展了反应的适用范围。[3]
均相反应条件
利用可溶性的O,O'-二(2-氨基乙基)聚乙烯醇(diamoPEG)作起始物,可以制得四氢呋喃可溶的三元聚合物。利用这种催化剂可以在均相条件下进行环氧化反应。[4]
相转移助催化
通过加入四丁基溴化铵作为相转移催化剂,可以大大提高反应速率。这是因为相转移催化剂的加入可以提高有机相中过氧化氢阴离子的浓度。[14]最初这一改进方法被用于两相反应模式,但对三相反应同样起作用。[5][12]
大规模生产
在生产中,固定相催化剂已被用在膜式反应器里,催化环氧化反应。目前正在开展将其应用于固定床连续反应器的研究。[11]
合成实例

(+)-黄皮内酰胺的全合成
Cappi等人利用朱莉娅-科隆纳环氧化反应,以固定在聚乙二醇上的聚L-亮氨酸为催化剂,DABCO-过氧化氢复合物(DABCO-H2O2) 或过氧化氢-尿素复合物为氧化剂,在微型固定床连续反应器中完成了全合成的一步。(图 7) 该实际应用也从概念上证明了,均相朱莉娅-科隆纳环氧化反应是可行的。[15]
-Clausenamide7.png.webp)
(+)-goniotriol 7, (+)-goniofufurone 8, (+)-8-acetylgoniotriol 9 和 gonio-pypyrone的全合成
Chen等人利用两相朱莉娅-科隆纳环氧化反应,以聚L-亮氨酸作催化剂,尿素-过氧化氢复合物作氧化剂,DBU作碱,完成了一系列从哥那香中提取的內酯的全合成路线中的关键一步。(图 8)这些內酯包括(+)-goniotriol 7, (+)-goniofufurone 8, (+)-8-acetylgoniotriol 9 和 gonio-pypyrone。[16]
-goniotriol.png.webp)
参考文献
- Juliá, Sebastián; Masana, Jaume; Vega, Juan Carlos. . Angewandte Chemie International Edition in English (Wiley-Blackwell). 1980, 19 (11): 929–931. ISSN 0570-0833. doi:10.1002/anie.198009291.
- Juliá, Sebastián; Guixer, Joan; Masana, Jaume; Rocas, José; Colonna, Stefano; Annuziata, Rita; Molinari, Henriette. . J. Chem. Soc., Perkin Trans. 1. 1982: 1317. doi:10.1039/P19820001317.
- Allen, Joanne V.; Bergeron, Sophie; Griffiths, Matthew J.; Mukherjee, Shubhasish; Roberts, Stanley M.; Williamson, Natalie M.; Wu, L. Eduardo. . J. Chem. Soc., Perkin Trans. 1. 1998, (19): 3171. doi:10.1039/A805407J.
- Flood, Robert W.; Geller, Thomas P.; Petty, Sarah A.; Roberts, Stanley M.; Skidmore, John; Volk, Martin. . Org. Lett. 2001, 3 (5): 683. doi:10.1021/ol007005l.
- Carrea, G; Colonna, S; Kelly, D; Lazcano, A; Ottolina, G; Roberts, S. . Trends in Biotech. 2005, 23 (10): 507. doi:10.1016/j.tibtech.2005.07.010.
- Kelly, David R.; Meek, Alastair; Roberts, Stanley M. . Chem. Comm. 2004, (18): 2021. doi:10.1039/B404379K.
- Kelly, D. R.; Roberts, S. M., The mechanism of polyleucine catalysed asymmetric epoxidation". Chem. Comm. 2004, (18), 2018-2020. doi:10.1039/B404390C
- Carrea, G.; Colonna, S.; Meek, A. D.; Ottolina, G.; Roberts, S. M., "Kinetics of chalcone oxidation by peroxide anion catalysed by poly-L-leucine". Chem. Comm. 2004, (12), 1412-1413. doi:10.1039/B401497A
- Berkessel, A.; Gasch, N.; Glaubitz, K.; Koch, C., "Highly enantioselective enone epoxidation catalyzed by short solid phase-bound peptides: Dominant role of peptide helicity". Org. Lett. 2001, 3 (24), 3839–3842. doi:10.1021/ol0166451
- Bentley, P. A.; Cappi, M. W.; Flood, R. W.; Roberts, S. M.; Smith, J. A., Towards a mechanistic insight into the Julia-Colonna asymmetric epoxidation of α,β-unsaturated ketones using discrete lengths of poly-leucine. Tet. Lett. 1998, 39 (50), 9297–9300. doi:10.1016/S0040-4039(98)02090-5
- Adger, B. M.; Barkley, J. V.; Bergeron, S.; Cappi, M. W.; Flowerdew, B. E.; Jackson, M. P.; McCague, R.; Nugent, T. C.; Roberts, S. M., "Improved procedure for Julia–Colonna asymmetric epoxidation of α,β-unsaturated ketones: total synthesis of diltiazem and Taxol (TM) side-chain". J. Chem. Soc.-Perkin Trans. 1 1997, (23), 3501–3507. doi:10.1039/A704413E
- Lopez-Pedrosa, J. M.; Pitts, M. R.; Roberts, S. M.; Saminathan, S.; Whittall, J., "Asymmetric epoxidation of some arylalkenyl sulfones using a modified Julia–Colonna procedure". Tet. Lett. 2004, 45 (26), 5073–5075. doi:10.1016/j.tetlet.2004.04.190
- Yi, H.; Zou, G.; Li, Q.; Chen, Q.; Tang, J.; He, M. Y., "Asymmetric epoxidation of alpha,beta-unsaturated ketones catalyzed by silica-grafted poly-(L)-leucine catalysts". Tet. Lett. 2005, 46 (34), 5665–5668. doi:10.1016/j.tetlet.2005.06.096
- Geller, T.; Gerlach, A.; Kruger, C. M.; Militzer, H. C., "Novel conditions for the Julia–Colonna epoxidation reaction providing efficient access to chiral, nonracemic epoxides". Tet. Lett. 2004, 45 (26), 5065–5067. doi:10.1016/j.tetlet.2004.04.188
- Cappi, M. W.; Chen, W. P.; Flood, R. W.; Liao, Y. W.; Roberts, S. M.; Skidmore, J.; Smith, J. A.; Williamson, N. M., "New procedures for the –Colonna asymmetric epoxidation: synthesis of (+)-clausenamide". Chem. Comm. 1998, (10), 1159-1160. doi:10.1039/A801450G
- Chen, W. P.; Roberts, S. M., "Julia–Colonna asymmetric epoxidation of furyl styryl ketone as a route to intermediates to naturally-occurring styryl lactones". J. Chem. Soc.-Perkin Trans. 1 1999, (2), 103–105. doi:10.1039/A808436J