格林-巴利综合征
格林-巴利症候群[1](,)又稱脫髓鞘多發性神經炎,是一種因免疫系統損害周邊神經系統,引起急性、發炎性、脱髓鞘性、多發性神經根神經炎,進而導致急性肌肉癱瘓的疾病[2]。本病的典型初始症狀為痛覺異常及肌肉弱化。一般肌肉症狀會先從手腳開始進犯[2],之後進犯上臂及上半身[2]。本病症狀會持續數小時至數週[2],在急性期間,15%的患者會進犯呼吸肌,可能危及生命,需要使用機械式呼吸輔助[3],有些則會影響自律神經系統,導致心率及血壓異常[2]。
此疾病的致病原因尚未明朗[2],病理的機制和自體免疫性疾病有關,身體內的免疫系统攻擊週邊神經,因此破壞了髓磷脂的絕緣[2]。有時這種免疫失調會因為感染所引發,偶爾也會因為手術或疫苗接種所引發[2][3]。此疾病一般會依症狀及體徵診斷,不過需排除其他可能的病因,也會透過神經傳導研究測試以及檢查腦脊液來驗證[2]。疾病可依照虛弱的部位、神經傳導測試的結果、以及是否有對抗醣脂質抗體可以再分為幾個子類[4]。格林-巴利症候群屬於急性的多發性神經病[3]。
對於肌肉極度無力的病患,立即靜脈注射免疫球蛋白、進行血漿置換術,同時施以支持療法可有效使其中大部分恢復[2],患者的恢復期需數週到幾年之久[2],約三分之一的患者會留有終生的肌肉無力的後遺症[2]。格林-巴利症候群是一種罕見疾病,發生率約為每年十萬分之二[2][5],其全球死亡率約為7.5%[3]。性別與地域與其發生率不存在明顯關聯[2][3]。格林-巴利症候群以法國神經病學醫師若尔日·吉兰(Georges Guillain)與讓·亞歷山大·巴雷(Jean Alexandre Barré)的姓氏命名,兩位醫師與安德烈·斯托爾(André Strohl)醫師在1916年共同描述該疾病[6][7]。
症状和体征
此病最初的典型表现为感觉异常、麻木、麻刺、刺痛、疼痛,症状首现于四肢远端(手足),之后症状常常在数小时至数周内不断向近端进展且发生肢体肌肉无力,躯干、头部(尤舌头、口周)也会出现症状,症状一般两侧对称。[2]在疾病的急性期,15%患者可能因为呼吸肌受累而有生命危险,需要機械式呼吸輔助。本病除累及感觉神经、运动神经,导致上述典型症状外,还有一些患者可能有自主神经系统受累,导致诸如血压异常、心率异常等症状,也可能导致患者的死亡。[2]
病因和诊断
本病通常被认为是自體免疫性疾病,由人体的免疫系统错误地攻击自身周围神经的髓鞘,导致患者周围神经系统出现脱髓鞘损害。有的患者还会出现轴索损害。[2]本病的病因尚不明确。患者的免疫系统紊乱有时由感染诱发,由手术或疫苗诱发的少见,还有很多病人无明显诱发因素。[2][3][8]本病的诊断一般基于症状和体征,要排除其他疾病的可能,并有诸如肌电图神经传导检测、脑脊液检查等辅助检查证据支持。[2]
临床亚型
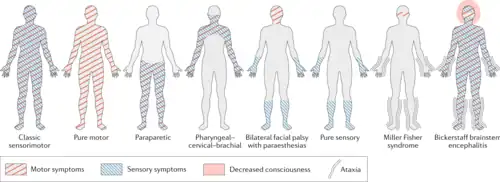
依据肌无力位置、肌电图结果、抗神经节苷脂抗体检测结果等,格林-巴利症候群可分为若干不同的亚型,每种亚型都有各自不同的特点。[4]
| 亚型[4] | 症状 | 流行病学 | 肌电图结果 | 抗神经节苷脂抗体 |
|---|---|---|---|---|
| 急性炎症性脱髓鞘性多发性神经病(AIDP) | 感觉症状、肌无力,常伴颅神经和自主神经系统受累 | 欧洲和北美最常见的亚型 | 脱髓鞘、多发性神经病 | 无明确证据 |
| 急性运动性轴索神经病(AMAN) | 肌无力,一般不伴感觉症状;颅神经受累罕见 | 欧美少见,而在亚洲、中南美洲,占全部GBS患者数量的30-65%,因而有时被叫做“中国麻痹综合征” | 轴索损害为主的多发性神经病,感觉动作电位一般正常 | GM1a/b, GD1a & GalNac-GD1a |
| 急性运动感觉轴索神经病(AMSAN) | 与AMAN相似,但伴有感觉症状 | - | 轴索损害为主的多发性神经病,感觉动作电位减少或缺失 | GM1, GD1a |
| Pharyngeal-cervical-brachial变异型 | 喉、面、颈、肩的肌肉明显无力 | - | 大体正常,仅有上肢局部的轴索损害 | 多见GT1a,偶有GQ1b,少见GD1a |
| Miller Fisher综合征 | 共济失调,眼肌无力,反射异常,但通常无肢体虚弱 | 男多于女,男女比2:1。常常出现于春季,患者多数大于43岁[9] | 大体正常,感觉传导有时有离散的变化或检测到H-reflex | GQ1b, GT1a |
治疗
出现严重无力症状的患者,应当接受静注免疫球蛋白或血浆置换治疗以封闭自體免疫抗体。同时还应当接受机械通气等对症支持治疗。虽然病情迅速进展且凶险,不接受正规治疗死亡率高,但经过正规治疗的患者预后往往较好,可在数周至数年内康复,但大约三分之一的患者可能会留有永久性的无力症状。[2]
流行病学
全球范围内,本病患者的死亡率大约为7.5%,[2][3][5]而发病率则为每年十万分之一到二之间,男女之间、各国各地区之间的发病率没有明显差异。[2][3]本病由法国神经病学家Georges Guillain、Jean Alexandre Barré、André Strohl在1916年首次描述,并依据前两位的姓命名为格林-巴利症候群。[6][7]
参考資料
- . [2021-09-09]. (原始内容存档于2020-12-04).
- . NIAMS. June 1, 2016 [13 August 2016]. (原始内容存档于5 August 2016).
- Ferri, Fred F. . Elsevier Health Sciences. 2016: 529. ISBN 9780323448383. (原始内容存档于2016-08-21) (英语).
- van den Berg, Bianca; Walgaard, Christa; Drenthen, Judith; Fokke, Christiaan; Jacobs, Bart C.; van Doorn, Pieter A. . Nature Reviews Neurology. 15 July 2014, 10 (8): 469–482. PMID 25023340. doi:10.1038/nrneurol.2014.121.
- Sejvar, James J.; Baughman, Andrew L.; Wise, Matthew; Morgan, Oliver W. . Neuroepidemiology. 2011, 36 (2): 123–133 [2017-04-01]. PMID 21422765. doi:10.1159/000324710. (原始内容存档于2014-12-04).
- van Doorn, Pieter A; Ruts, Liselotte; Jacobs, Bart C. . The Lancet Neurology. October 2008, 7 (10): 939–950. PMID 18848313. doi:10.1016/S1474-4422(08)70215-1.
- Eldar AH, Chapman J. . Autoimmunity Reviews. April 2014, 13 (4–5): 525–30. PMID 24434363. doi:10.1016/j.autrev.2014.01.033.
- FAQ:認識吉巴氏綜合症 (页面存档备份,存于)明報2010-01-08
- Mori M, Kuwabara S, Yuki N. . Expert Rev Neurother. January 2012, 12 (1): 39–51. PMID 22149656. doi:10.1586/ern.11.182.