氮化矽
氮化硅是由硅元素和氮元素构成的化合物。在氮气气氛下,将单质硅的粉末加热到1300-1400°C之间,硅粉末样品的重量随着硅单质与氮气的反应递增。在没有铁催化剂的情况下,约7个小时后硅粉样品的重量不再增加,此时反应完成生成Si3N4。除了Si3N4外,还有其他几种硅的氮化物(根据氮化程度和硅的氧化态所确定的相对应化学式)也已被文献所报道。比如气态的一氮化二硅(Si2N)、一氮化硅(SiN)和三氮化二硅(Si2N3)。这些化合物的高温合成方法取决于不同的反应条件(比如反应时间、温度、起始原料包括反应物和反应容器的材料)以及纯化的方法。[2]
| 氮化矽 | |
|---|---|
 | |
| IUPAC名 Silicon nitride | |
| 识别 | |
| CAS号 | 12033-89-5 |
| PubChem | 3084099 |
| ChemSpider | 2341213 |
| SMILES |
|
| InChI |
|
| InChIKey | HQVNEWCFYHHQES-UHFFFAOYSA-N |
| EINECS | 234-796-8 |
| MeSH | Silicon+nitride |
| 性质 | |
| 化学式 | Si3N4 |
| 外观 | 灰色无味粉末 |
| 密度 | 3.2 g/cm3, 固体 |
| 熔点 | 1900 °C(2173 K)(分解) |
| 折光度n D |
2.016[1] |
| 危险性 | |
| 欧盟分类 | 未列出 |
| 相关物质 | |
| 其他阴离子 | 碳化硅 二氧化硅 |
| 其他阳离子 | 氮化硼 |
| 若非注明,所有数据均出自标准状态(25 ℃,100 kPa)下。 | |
Si3N4是硅的氮化物中化学性质最为稳定的(仅能被稀的HF和热的H3PO4分解),也是所有硅的氮化物中热力学最稳定的。所以一般提及“氮化硅”时,其所指的就是Si3N4。它也是硅的氮化物中最重要的化合物商品。
在很宽的温度范围内氮化硅都是一种具有一定的热导率、低热膨胀系数、弹性模量较高的高强度硬陶瓷。不同于一般的陶瓷,它的断裂韧性高。这些性质结合起来使它具有优秀的耐热冲击性能,能够在高温下承受高结构载荷并具备优异的耐磨损性能。常用于需要高耐用性和高温环境下的用途,诸如气轮机、汽车引擎零件、轴承和金属切割加工零件。美国国家航空航天局的航天飞机就是用氮化硅制造的主引擎轴承。氮化硅薄膜是硅基半导体常用的绝缘层,由氮化硅制作的悬臂是原子力显微镜的传感部件。
历史
亨利·爱丁·圣克莱尔·德维尔和弗里德里希·维勒在1857年首次报道了氮化硅的合成方法。[3]在他们报道的合成方法中,为减少氧气的渗入而把另一个盛有硅的坩埚埋于一个装满碳的坩埚中加热。他们报道了一种他们称之为硅的氮化物的产物,但他们未能弄清它的化学成分。1879年Paul Schuetzenberger通过将硅与衬料(一种可作为坩埚衬里的糊状物,由木炭、煤块或焦炭与粘土混合得到)混合后在高炉中加热得到的产物,并把它报道为成分是Si3N4的化合物。1910年路德维希·魏斯和特奥多尔·恩格尔哈特在纯的氮气下加热硅单质得到了Si3N4。[4]1925年Friederich和Sittig利用碳热还原法在氮气气氛下将二氧化硅和碳加热至1250-1300°C合成氮化硅[5]
在后来的数十年中直到应用氮化硅的商业用途出现前,氮化硅未受到重视和研究。从1948年至1952年期间,艾奇逊开办在纽约州尼亚加拉大瀑布附近的金刚砂公司为氮化硅的制造和使用注册了几项专利。[6]1958年联合碳化物公司生产的氮化硅被用于制造热电偶管、火箭喷嘴和熔化金属所使用的坩埚。英国对氮化硅的研究工作始于1953年,目的是为了制造燃气涡轮机的高温零件。由此使得键合氮化硅和热压氮化硅得到发展。1971年美国国防部下属的国防高等研究计划署与福特和西屋公司签订一千七百万美元的合同研制两种陶瓷燃气轮机。[7]
虽然氮化硅的特性早已广为人知,但在地球自然界中存在的氮化硅(大小约为2×5µm)还是在二十世纪90年代才在陨石中被发现。为纪念质谱研究的先驱阿尔弗雷德·奥托·卡尔·尼尔将自然界中发现的此类氮化硅矿石冠名为“nierite”。[8]不过有证据显示可能在更早之前就在前苏联境内的阿塞拜疆发现过这种存在于陨石中的氮化硅矿石。[9]。含有氮化硅矿物的陨石也曾在中国贵州省境内发现过。[10]除存在于地球上的陨石中以外,氮化硅也分布于外层空间的宇宙尘埃中。[11]
合成方法
可在1300-1400°C的条件下用单质硅和氮气直接进行化合反应得到氮化硅: [6]
- 3 Si(s) + 2 N
2(g) → Si
3N
4(s)
- SiCl
4(l) + 6 NH
3(g) → Si(NH)
2(s) + 4 NH
4Cl(s) 在0 °C的条件下 - 3 Si(NH)
2(s) → Si
3N
4(s) + N
2(g) + 3 H
2(g) 在1000 °C的条件下
或用碳热还原反应在1400-1450°C的氮气气氛下合成:[6]
- 3 SiO
2(s) + 6 C(s) + 2 N
2(g) → Si
3N
4(s) + 6 CO(g)
对单质硅的粉末进行渗氮处理的合成方法是在二十世纪50年代随着对氮化硅的重新“发现”而开发出来的。也是第一种用于大量生产氮化硅粉末的方法。但如果使用的硅原料纯度低会使得生产出的氮化硅含有杂质硅酸盐和铁。用二胺分解法合成的氮化硅是无定形态的,需要进一步在1400-1500°C的氮气下做退火处理才能将之转化为晶态粉末,目前二胺分解法在重要性方面是仅次于渗氮法的商品化生产氮化硅的方法。碳热还原反应是制造氮化硅的最简单途径也是工业上制造氮化硅粉末最符合成本效益的手段。[6]
电子级的氮化硅薄膜是通过化学气相沉积或者等离子体增强化学气相沉积技术制造的:[6][12]
- 3 SiH
4(g) + 4 NH
3(g) → Si
3N
4(s) + 12 H
2(g) - 3 SiCl
4(g) + 4 NH
3(g) → Si
3N
4(s) + 12 HCl(g) - 3 SiCl
2H
2(g) + 4 NH
3(g) → Si
3N
4(s) + 6 HCl(g) + 6 H
2(g)
如果要在半导体基材上沉积氮化硅,有两种方法可供使用:[12]
- 利用低压化学气相沉积技术在相对较高的温度下利用垂直或水平管式炉进行。[13]
- 等离子体增强化学气相沉积技术在温度相对较低的真空条件下进行。
氮化硅的晶胞参数与单质硅不同。因此根据沉积方法的不同,生成的氮化硅薄膜会有产生张力或应力。特别是当使用等离子体增强化学气相沉积技术时,能通过调节沉积参数来减少张力。[14]
先利用溶胶凝胶法制备出二氧化硅,然后同时利用碳热还原法和氮化对其中包含特细碳粒子的硅胶进行处理后得到氮化硅纳米线。硅胶中的特细碳粒子是由葡萄糖在1200-1350°C分解产生的。合成过程中涉及的反应可能是:[15]
- SiO
2(s) + C(s) → SiO(g) + CO(g) - 3 SiO(g) + 2 N
2(g) + 3 CO(g) → Si
3N
4(s) + 3 CO
2(g) 或 - 3 SiO(g) + 2 N
2(g) + 3 C(s) → Si
3N
4(s) + 3 CO(g)
加工方法
作为粒状材料的氮化硅是很难加工的——不能把它加热到它的熔点1850°C以上,因为超过这个温度氮化硅发生分解成硅和氮气。因此用传统的热压烧结技术是有问题的。把氮化硅粉末粘合起来可通过添加一些其他物质比如烧结助剂或粘合剂诱导氮化硅在较低的温度下发生一定程度的液相烧结后粘合成块状材料。[16]但由于需要添加粘合剂或烧结助剂,所以这种方法会在制出的块状材料中引入杂质。使用放电等离子烧结是另一种可以制备更纯净大块材料的方法,对压实的粉末在非常短的时间内(几秒中)进行电流脉冲,用这种方法能在1500-1700°C的温度下得到紧实致密的氮化硅块状物。[17][18]
晶体结构和特性
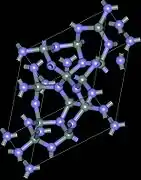
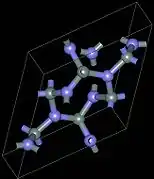
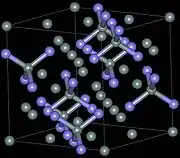
氮化硅(Si3N4)存在有3种结晶结构,分别是α、β和γ三相。α和β两相是Si3N4最常出现的型式,且可以在常压下制备。γ相只有在高压及高温下,才能合成得到,它的硬度可达到35GPa[19],为包含八面体形六配位硅原子的尖晶石型结构。
参考资料
- Refractive index database. refractiveindex.info
- O. N. Carlson. . Bulletin of Alloy Phase Diagrams. 1990-12-01, 11 (6): 569–573 [2013-02-06]. doi:10.1007/BF02841719. (原始内容存档于2015-04-26).
- Deville, H. and Wohler, F. . Liebigs Ann. Chem. 1857, 104: 256 [2013-02-06]. doi:10.1002/jlac.18571040224. (原始内容存档于2015-04-14).
- Ludwig Weiss, Theodor Engelhardt. . Z. Anorg. Allgem. Chem. 1910, 1910 (1): 38–104 [2013-02-06]. doi:10.1002/zaac.19090650107. (原始内容存档于2016-03-16).
- Ernst Friederich, Lieselotte Sittig. . Z. Anorg. Allgem. Chem. 1925, 143: 293–320 [2013-02-06]. doi:10.1002/zaac.19251430121. (原始内容存档于2016-03-05).
- Riley, Frank L. . Journal of the American Ceramic Society. 2004, 83 (2): 245-265 [2013-02-06]. doi:10.1111/j.1151-2916.2000.tb01182.x. (原始内容存档于2012-12-07).
- Carter, C. Barry and Norton, M. Grant. . Springer. 2007: 27. ISBN 0-387-46270-8.
- Lee, M. R.; Russell, S. S.; Arden, J. W.; Pillinger, C. T. . Meteoritics. July 1995, 30 (4): 387–398 [2013-02-06]. Bibcode:1995Metic..30..387L. doi:10.1111/j.1945-5100.1995.tb01142.x. (原始内容存档于2016-03-07).
- Igor V. Petkov. 32. Ocean Picture Ltd. : 719–732.
- Lin, Yangting, Amari, Sachiko, and Pravdivtseva, Olga. . The Astrophysical Journal. 2002-08, 525 (1): 257–263. doi:10.1086/341218.
- Jones, Anthony P. . European Journal of Mineralogy. 2007-12-17, 19 (6): 771–782 [2013-02-07]. doi:10.1127/0935-1221/2007/0019-1766. (原始内容存档于2014-03-06).
- Yoshio Nishi, Robert Doering. . CRC Press. 2000: 324–325. ISBN 0-8247-8783-8.
- . [2009-06-06]. (原始内容存档于2009-06-05).
- . [2009-06-06]. (原始内容存档于2009-06-05).
- Ghosh Chaudhuri, Mahua; Dey, Rajib; Mitra, Manoj K; Das, Gopes C; Mukherjee, Siddhartha. . Sci. Technol. Adv. Mater. 2008, 9 (1): 015002. Bibcode:2008STAdM...9a5002G. doi:10.1088/1468-6996/9/1/015002.
- . [2009-06-06]. (原始内容存档于2009-01-07).
- Nishimura, Toshiyuki; Xu, Xin; Kimoto, Koji; Hirosaki, Naoto; Tanaka, Hidehiko. . Sci. Technol. Adv. Mater. 2007, 8 (7–8): 635. Bibcode:2007STAdM...8..635N. doi:10.1016/j.stam.2007.08.006.
- Peng, H. . Stockholm University. 2004 [2009-06-06]. (原始内容存档于2011-05-12).
- J Z Jiang, F Kragh, D J Frost, K Ståhl and H Lindelov. . J. Phys.: Condens. Matter. 4 June 2001, 13 (22): 515. doi:10.1088/0953-8984/13/22/111.