能带结构
为何有能带
单獨原子的电子占据了原子轨道,形成一个分立的能级结构。如果几个原子集合成分子,他们的原子轨道发生类似于耦合振荡的分离。这会产生与原子数目成比例的分子轨道。当数目非常巨大(数量级为或更多)的原子集合成固体时,轨道数目急剧增多,轨道相互间的能量的差别将会变的非常小。虽然,无论多少原子聚集在一起,轨道能量的分布都是不连续的,但因为固体中的能级如此之多,所以一群相邻的能级,可以被视为一个连续能谱,也就是一个能带。而能带与能带之间的间隙,则为能隙。

物理学中流行的方法是从不带电的电子和原子核出发,因为它们是自由的平面波,可以具有任意能量,并在带电后衰减。这导致了布拉格反射和能带结构。
基本概念
晶体结构的对称性与波向量
晶体能带结构的计算需要利用晶格的周期性和对称性 ,通常会假设晶体是理想的 ,并具有周期边界和离散的平移对称性。单电子薛定谔方程在晶体的周期性势垒中的解即为布洛赫波,如下所示:
- ,

其中k称为波向量。对于每一个k,薛定谔方程都有多个解,解的数量用n表示,也就是能带的条数。每条能带都随着k周期性变化,我们用En(k)来表示。对于包含原子数非常巨大的真实晶体,可以当成趋于热力学极限并恢复平移对称性,并用布洛赫波近似地描述晶体的体态波函数性质。
由于晶体结构的离散平移对称性,能带图通常只画第一布里渊区以内的k。第一布里渊区之外的波向量对应的解和之内的解是对应的。
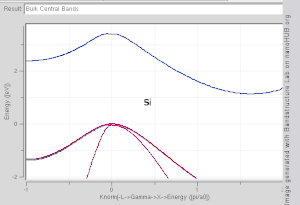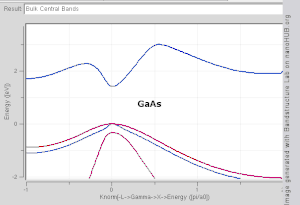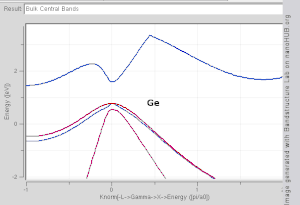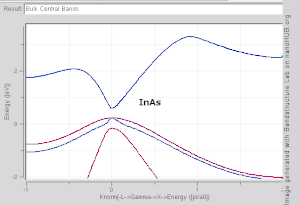
由于布里渊区中高对称点一般会伴随能级简并 ,并反应晶格对称性,所以通常会沿着高对称点来展示能带结构图。 布里渊区中的高对称点常常被记为Γ, Δ, Λ, Σ。此处展示几种常见半导体的能带结构图如右图所示。
不同固体的能带结构

固体材料的能带结构由多条能带组成,能带分为导带、价带和禁带等,导带和价带间的空隙称为禁带(能隙)(即右边第二副图中所示的)。

能带结构可以解释固体中导体、半导体、绝缘体三大类区别的由来。材料的导电性是由“传导带”中含有的电子数量决定。当电子从“价带”获得能量而跳跃至“传导带”时,电子就可以在带间任意移动而导电。
一般常见的金属导体,因为其传导带与价带之间的“能隙”非常小,在室温下电子很容易获得能量而跳跃至传导带而导电,而绝缘材料(绝缘体)则因为能隙很大(通常大于9电子伏特),电子很难跳跃至传导带,所以无法导电。一般半导体材料的能隙约为1至3电子伏特,介于导体和绝缘体之间。因此只要给予适当条件的能量激发,或是改变其能隙之间距,此材料就能导电。
导带的最低点和价带的最高点处于同一k位置的称为直接带隙半导体。比如GaAs和InAs。 导带的最低点和价带的最高点处于不同k位置的称为间接带隙半导体。比如Si和Ge。他们的能带结构图如右上第一幅图所示。
态密度
态密度函数g(E)是指电子在能量E处单位体积单位能量的电子态数量。
态密度函数对于在能带理论基础上相关的计算非常重要。例如,g(E)乘以费米-狄拉克分布即可以得到电子浓度与能量的关系式。
在能带间隙中,态密度g(E)=0。而在范霍夫奇点(Van Hove singularity),態密度会是發散的。
能带填充
在热力学平衡状态下,能量E充满电子的可能性由费米-狄拉克(Fermi-Dirac)分布给出,该热力学分布考虑了泡利不相容原理。 尽管有无限多个能带,因此有无限多个状态,但是在这些能带中只有有限数量的电子。电子数的优选值是静电的结果:即使材料的表面可以带电,材料的内部体积也倾向于是电荷中性的。电荷中性的条件意味着N / V必须与材料中质子的密度匹配。为此,材料会进行静电自我调节,使能带结构上下移动能量(从而移动g(E)),直到相对于费米能级达到正确的平衡。
费米能级附近的能带名称(导带,价带) 固体具有无限数量的允许带,就像原子具有无限多个能级一样。但是,大多数能带的能量太高,通常在通常情况下会被忽略。[1]相反,与核心轨道相关的能带非常低(例如1s电子)。这些低能芯带通常也被忽略,因为它们始终保持充满电子,因此是惰性的。[2]同样,材料在其整个能带结构中都具有多个带隙。
与电子和光电相关的最重要的带和带隙是那些能量接近费米能级的带。根据材料,费米能级附近的带和带隙被赋予特殊名称:
在半导体或带绝缘子中,费米能级被带隙所围绕,称为带隙(以将其与带结构中的其他带隙区分开)。带隙以上的最接近的带称为导带,带隙下方的最接近的带称为价带。 “价带”之类的名称类似于化学,因为在半导体(和绝缘体)中,价带是由价轨道构成的。 在金属或半金属中,费米能级在一个或多个允许带的内部。在半金属中,该带通常被称为“导带”或“价带”,这取决于电荷传输是更类似于电子的还是类似于空穴的,类似于半导体。然而,在许多金属中,这些带既不像电子一样也不像空穴一样,通常是由价态轨道组成的,因此通常被称为“价带”。[3] 金属的能带结构中的能隙对低能物理并不重要,因为它们与费米能级相距太远。
晶体的能带结构
晶体能带结构理论
拟设是使用布洛赫波的周期性晶格中电子波的特例,通常在衍射动力学理论中对其进行处理。每个晶体都是可以用Bravais晶格表征的周期性结构,对于每个Bravais晶格,我们可以确定倒易晶格,该倒易晶格将周期性封装在三个倒易晶格矢量(b1,b2,b3)的集合中。现在,可以将与直接晶格共享相同周期性的任何周期性电势V(r)展开为傅立叶级数,该傅立叶级数的唯一不消失的成分是与倒易晶格矢量相关的成分。
近自由电子近似
在近自由电子的近似中,电子之间的相互作用被完全忽略。这种近似允许使用布洛赫定理,布洛赫定理指出周期性电势中的电子具有波函数和能量,这些波函数和能量在波矢量中具有周期性,直到相邻倒易晶格矢量之间的相移恒定为止。 近自由电子近似模型在金属(如相邻原子之间的距离很小)的材料计算中效果特别好。在这种材料中,原子轨道和相邻原子上的电势的重叠相对较大。在那种情况下,电子的波函数可以通过(修正的)平面波来近似。像铝这样的金属的能带结构甚至接近于空晶格近似。
紧束缚模型
与近自由电子近似相反的极端假设晶体中的电子的行为非常像组成原子的集合。这个紧束缚模型假定独立的单电子Schrödinger方程的解可以通过原子轨道的线性组合很好地近似. 紧束缚模型的近似在原子轨道与相邻原子上的电势重叠有限的材料中效果很好。紧束缚模型-哈密顿主义者基于原子sp3轨道很好地描述了诸如Si,GaAs,SiO2和金刚石之类的材料的能带结构。在过渡金属中,混合的紧束缚-近自由电子近似模型用于描述宽的近自由电子近似导带和窄的嵌入式紧束缚 d带。 Wannier函数的原子轨道部分的径向函数最容易通过使用伪电势方法来计算。 近自由电子近似模型,紧束缚模型或组合的近自由电子近似-紧束缚模型频带结构计算[4]有时使用基于伪势方法的波动函数近似扩展,通常被用作进行进一步计算的起点。
KKR模型
Korringa-Kohn-Rostoker(KKR)方法最初是由Korringa在1947年开发的,他使用多重散射理论来求解Schrödinger方程并确定周期性固体的能带结构。七年后,Kohn和Rostoker 提出了一种与Korringa等效的方法,但使用了格林函时称之为Korringa-Kohn-Rostoker(KKR)方法。[5][6] KKR方法最重要的特征可能是,从非相对论描述转向相对论描述时,其形式结构不会改变。KKR 或格林函数公式最重要的特征是(1)它将问题的两个方面分开:结构(原子的位置)与散射(原子的化学特性); (2)格林函数提供了一种对电子特性进行局部描述的自然方法,可以适用于合金和其他无序系统。自四十年代后期以来,KKR方法已经进行了几处改进。这些改进之一是所谓的遮蔽(screened or tight binding)的Korringa-Kohn-Rostoker(sKKR)方法。 sKKR方法允许在多重散射理论的框架内以二维方式计算表面,界面以及通常具有二维周期性的一般分层系统的电子特性,而无需任何进一步的近似。这种近似的最简单形式是将非重叠球(称为松饼罐)置于原子位置上。在这些区域内,电子所经历的电势近似于关于给定的原子核是球对称的。在剩余的间隙区域中,屏蔽电位近似为常数。强制以原子为中心的球体和间隙区域之间的电势连续性。利用这种格林函数和Korringa–Kohn–Rostoker方法,可以获得Korringa–Kohn–Rostoker相干势近似(KKR-CPA)。 KKR-CPA用于计算替代固溶合金的电子态,而Bloch定理不成立。可以使用局部自洽多重散射(LSMS)方法找到更广泛范围的凝聚态结构的电子态,该方法也基于单粒子格林函数。
密度泛函理论
在最近的物理学文献中,大多数电子结构和能带图都是使用密度泛函理论(DFT)计算的,密度泛函理论不是模型而是一种理论,即凝聚态物理的微观第一原理理论,通过在电子密度的函数中引入交换相关项来解决电子-电子多体问题。在许多情况下,例如通过角度分辨光发射光谱法(ARPES),发现DFT计算的谱带与实验测量的谱带一致。特别地,带形通常通过DFT很好地再现。但是,与实验结果相比,DFT谱带也存在系统误差。尤其是,DFT似乎系统地低估了绝缘体和半导体中的带隙约30-40%。[7]
通常认为,DFT是仅预测系统基态性质(例如,总能量,原子结构等)的理论,并且激发态性质不能由DFT确定。这是一个误解。原则上,DFT可以确定给定将基态密度映射到该特性的功能的系统的任何特性(基态或激发态)。这是Hohenberg–Kohn定理的本质。[8] 然而,在实践中,不存在将基态密度映射到材料内电子的激发能的已知功能。因此,在文献中被称为DFT能带图的是DFT Kohn-Sham能量的表示,即虚构的非相互作用系统Kohn-Sham系统的能量,根本没有物理解释。不可将Kohn-Sham电子结构与系统的真实准粒子电子结构相混淆,而且对于Hartree-Fock能量也没有Koopman定理对Kohn-Sham能量的持有,可以将其真正视为准粒子能量的近似值。因此,从原理上讲,基于Kohn-Sham的DFT并不是能带理论,即不是适合于计算能带和能带曲线的理论。原则上,基于时间的DFT可用于计算真实的能带结构,尽管在实践中这通常很困难。一种流行的方法是使用混合功能,其中包含Hartree-Fock精确交换的一部分。这大大改善了半导体的预测带隙,但对金属和宽带隙材料的可靠性较差。[9]
格林函数方法和从头算起GW近似
为了计算包括电子-电子相互作用多体效应的能带,人们可以诉诸所谓的格林函数方法。确实,对系统格林函数功能的了解不仅提供了基态(总能量),而且还提供了系统的激发态可观测值。格林函数的极点是准粒子能量,即固体的能带。一旦知道系统的自能量,就可以通过求解戴森方程来计算格林函数。对于像固体这样的真实系统,自能量是一个非常复杂的量,通常需要近似值来解决该问题。一种这样的近似是GW近似,从数学形式上称自能量为格林函数G与动态筛选的相互作用W的乘积Σ= GW。这种方法在解决能带图的计算时更相关(以及超出范围的数量(例如光谱函数),并且也可以完全从头算起。 GW近似值似乎提供了与实验一致的绝缘体和半导体的带隙,从而纠正了系统性DFT低估。
动力学平均场理论
Mott绝缘体是指在金属中,尽管还有空的能阶,但是电子间的相互作用太强,导致其它的电子没有办法在空的能阶中移动。所以尽管看起来跟金属一样,但是导电性质却像绝缘体。尽管近乎自由的电子近似能够描述电子能带结构的许多特性,但该理论的一个结果是,它预测了每个晶胞中相同数量的电子。如果电子数是奇数,则我们可以预期每个晶胞中都有一个不成对的电子,因此价带未被完全占据,从而使该材料成为导体。但是,每单位晶格中具有奇数个电子的材料(例如CoO)是绝缘体,与该结果直接冲突。这种材料被称为Mott绝缘体,并且需要包含详细的电子-电子相互作用(在能带理论中仅作为对晶体电势的平均影响来处理)以解释差异。哈伯德模型是一个可以包含这些相互作用的近似理论。可以在所谓的动力学平均场理论中以非扰动的方式对待它,该理论试图弥合几乎自由电子近似与原子极限之间的差距。然而,在形式上,状态在这种情况下不是不相互作用的,并且带结构的概念不足以描述这些情况。
其它
计算能带结构是理论固态物理学中的重要课题。除上述模型外,其他模型还包括:
- 空晶格近似:已划分为晶格的自由空间区域的“带结构”。
- k·p扰动理论是一种仅用几个参数就可以大致描述能带结构的技术。该技术通常用于半导体,并且模型中的参数通常是通过实验确定的。
- Kronig-Penney模型,一维矩形井模型,用于说明带形成。它虽然简单,但可以预测许多重要现象,但不是定量的。
- 哈伯德模型:
能带结构已被普遍化为复数的波矢量,从而产生了所谓的复能带结构,可决定了材料的许多周期性被破坏的特性,例如在表面、界面和缺陷处,都令人关注。
每个模型都很好地描述了某些类型的固体,而其他模型则描述得很差。几乎自由的电子模型对金属有效,但对非金属则较差。紧束缚模型对于离子绝缘(例如金属卤化物盐(例如NaCl))非常精确。
参考资料
- High-energy bands are important for electron diffraction physics, where the electrons can be injected into a material at high energies, see Stern, R.; Perry, J.; Boudreaux, D. . Reviews of Modern Physics. 1969, 41 (2): 275. Bibcode:1969RvMP...41..275S. doi:10.1103/RevModPhys.41.275..
- Low-energy bands are however important in the Auger effect.
- In copper, for example, the effective mass is a tensor and also changes sign depending on the wave vector, as can be seen in the de Haas–van Alphen effect; see https://www.phys.ufl.edu/fermisurface/ (页面存档备份,存于)
- Walter Ashley Harrison. . Dover Publications. 1989. ISBN 978-0-486-66021-9.
- Joginder Singh Galsin. . Springer. 2001. Appendix C. ISBN 978-0-306-46574-1.
- Kuon Inoue, Kazuo Ohtaka. . Springer. 2004: 66. ISBN 978-3-540-20559-3.
- Theoretical study on copper’s energetics and magnetism in TiO2 polymorphs (页面存档备份,存于) Journal of Applied Physics, 2013,113(23), 233913
- Hohenberg, P; Kohn, W. . Phys. Rev. Nov 1964, 136 (3B): B864–B871. Bibcode:1964PhRv..136..864H. doi:10.1103/PhysRev.136.B864.
- Paier, J.; Marsman, M.; Hummer, K.; Kresse, G.; Gerber, IC.; Angyán, JG. . J Chem Phys. Apr 2006, 124 (15): 154709. Bibcode:2006JChPh.124o4709P. PMID 16674253. doi:10.1063/1.2187006.
參考文獻
- Kotai no denshiron (The theory of electrons in solids), by Hiroyuki Shiba, ISBN 4-621-04135-5
- Microelectronics, by Jacob Millman and Arvin Gabriel, ISBN 0-07-463736-3, Tata McGraw-Hill Edition.
- Solid State Physics, by Neil Ashcroft and N. David Mermin, ISBN 0-03-083993-9,
- Introduction to Solid State Physics by Charles Kittel, ISBN 0-471-41526-X
- Electronic and Optoelectronic Properties of Semiconductor Structures - Chapter 2 and 3 by Jasprit Singh, ISBN 0-521-82379-X
参见
- 费米面
- 有效质量
- 固体物理学