
ATP7A, also known as Menkes' protein (MNK), is a copper-transporting P-type ATPase which uses the energy arising from ATP hydrolysis to transport Cu(I) across cell membranes. The ATP7A protein is a transmembrane protein and is expressed in the intestine and all tissues except liver. In the intestine, ATP7A regulates Cu(I) absorption in the human body by transporting Cu(I) from the small intestine into the blood. In other tissues, ATP7A shuttles between the Golgi apparatus and the cell membrane to maintain proper Cu(I) concentrations (since there is no free Cu(I) in the cell, Cu(I) ions are all tightly bound) in the cell and provides certain enzymes with Cu(I) (e.g. peptidyl-α-monooxygenase, tyrosinase, and lysyl oxidase). The X-linked, inherited, lethal genetic disorder of the ATP7A gene causes Menkes disease, a copper deficiency resulting in early childhood death.[5]
Gene
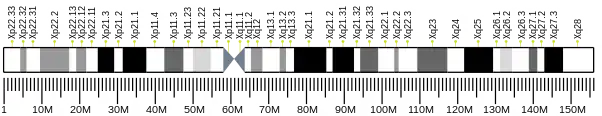
The ATP7A gene is located on the long (q) arm of the X chromosome at band Xq21.1. The encoded ATP7A protein has 1,500 amino acids.[6] At least 12 disease-causing mutations in this gene have been discovered. [7] Mutations/additions/deletions of this gene often cause copper deficiency, which leads to progressive neurodegeneration and death in children.[8]
Structure
ATP7A is a transmembrane protein with the N- and C-termini both oriented towards the cytosol (see picture). It is highly homologous to protein ATP7B. ATP7A contains three major functional domains:[9][10][11][12]
- Eight transmembrane segments that form a channel and allow for Cu(I) to pass through the membrane;
- An ATP-binding domain;
- A large N-terminal cytosolic domain that contains six repeated Cu(I)-binding sites, each containing a GMTCXXC motif.

Many motifs in the ATP7A structure are conserved:[11]
- The TGEA motif lies in the loop on the cytosolic side between transmembrane segments 4 and 5 and is involved in energy transfer.
- The CPC motif located in transmembrane segment 6 is common for all heavy metal transporting ATPases.
Between transmembrane segments 6 and 7 is a large cytoplasmic loop, where three motifs are located: DKTG, SEHPL, and GDGXND.
- The DKTG motif is essential for the proper function of the ATPase. The aspartic acid (D) residue is phosphorylated during the transport cycles.
- The SEHPL motif only exists in heavy metal transporting P-type ATPases. Without the histidine (H) residue ATP7A may not function properly.
- The GDGXND motif near transmembrane segment 7 is thought to contain mainly α-helices and serves as a structural support.
The six Cu(I)-binding sites at the N-terminal bind one Cu(I) each. This binding site is not specific for Cu(I) and can bind various transition metal ions. Cd(II), Au(III) and Hg(II) bind to the binding site more tightly than does Zn(II), whereas Mn(II) and Ni(II) have lower affinities relative to Zn(II). In the case of Cu(I), a possible cooperative-binding mechanism is observed. When the Cu(I) concentration is low, Cu(I) has a lower affinity for ATP7A compared to Zn(II); as the Cu(I) concentration increases, a dramatic increasing affinity of Cu(I) for the protein is observed.[11]
Conformational change
The two cysteine (C) residues in each Cu(I)-binding site are coordinated to Cu(I) with a S-Cu(I)-S angle between 120 and 180° and a Cu-S distance of 2.16 Å. Experimental results from a homologous protein ATP7B suggests that reducing reagents are involved, and upon Cu(I) binding the disulfide bonding between the cysteine residues is broken as cysteine starts to bind to Cu(I), leading to a series of conformational changes at the N-terminal of the protein, and possibly activating the Cu(I)-transporting activity of other cytosolic loops.[11]
Of the six copper(I)-binding sites, two are considered enough for the function of Cu(I) transport. The reason why there are six binding sites remains not fully understood. However, some scientists have proposed that the other four sites may serve as a Cu(I) concentration detector.[9]
Transport mechanism
ATP7A belongs to a transporter family called P-type ATPases, which catalyze auto-phosphorylation of a key conserved aspartic acid (D) residue within the enzyme. The first step is ATP binding to the ATP-binding domain and Cu(I) binding to the transmembrane region. Then ATP7A is phosphorylated at the key aspartic acid (D) residue in the highly conserved DKTG motif, accompanied by Cu(I) release. A subsequent dephosphorylation of the intermediate finishes the catalytic cycle. Within each cycle, ATP7A interconverts between at least two different conformations, E1 and E2. In the E1 state, Cu(I) is tightly bound to the binding sites on the cytoplasmic side; in the E2 state, the affinity of ATP7A for Cu(I) decreases and Cu(I) is released on the extracellular side.[13]
Function
ATP7A is important for regulating copper Cu(I) in mammals.[10] This protein is found in most tissues, but it is not expressed in the liver.[11] In the small intestine, the ATP7A protein helps control the absorption of Cu(I) from food. After Cu(I) ions are absorbed into enterocytes, ATP7A is required to transfer them across the basolateral membrane into the circulation.[9]
In other organs and tissues, the ATP7A protein has a dual role and shuttles between two locations within the cell. The protein normally resides in a cell structure called the Golgi apparatus, which modifies and transports newly produced enzymes and other proteins. Here, ATP7A supplies Cu(I) to certain enzymes (e.g. peptidyl-α-monooxygenase, tyrosinase, and lysyl oxidase[9]) that are critical for the structures and functions of brain, bone, skin, hair, connective tissue, and the nervous system. If Cu(I) levels in the cell environment are elevated, however, ATP7A moves to the cell membrane and eliminates excess Cu(I) from the cell.[8][10]
The functions of ATP7A in some tissues of the human body are as follows:[10]
| Tissue | Location | Function |
|---|---|---|
| Kidney | Expressed in epithelial cells of the proximal and distal renal tubules | Removes excess Cu(I) to maintain Cu(I) level in the kidney |
| Parenchyma | In the cytotrophoblast, syncytiotrophoblast and foetal vascular endothelial cells | Delivers Cu(I) to placental cuproenzymes and transports Cu(I) into the foetal circulation |
| Central nervous system | Various locations | Distributes Cu(I) in the various compartments of the central nervous system |
Interactions
ATP7A has been shown to interact with ATOX1 and GLRX. Antioxidant 1 copper chaperone (ATOX1) is required to maintain Cu(I) copper homeostasis in the cell. It can bind and transport cytosolic Cu(I) to ATP7A in the trans-Golgi-network. Glutaredoxin-1 (GRX1) has is also essential for ATP7A function. It promotes Cu(I) binding for subsequent transport by catalyzing the reduction of disulfide bridges. It may also catalyze de-glutathionylation reaction of the C (cysteine) residues within the six Cu(I)-binding motifs GMTCXXC.[10]
Clinical significance
Menkes disease is caused by mutations in the ATP7A gene.[14] Researchers have identified different ATP7A mutations that cause Menkes disease and occipital horn syndrome (OHS), the milder form of Menkes disease. Many of these mutations delete part of the gene and are predicted to produce a shortened ATP7A protein that is unable to transport Cu(I). Other mutations insert additional DNA base pairs or use the wrong base pairs, which leads to ATP7A proteins that do not function properly.[6]
The altered proteins that result from ATP7A mutations impair the absorption of copper from food, fail to supply copper to certain enzymes, or get stuck in the cell membrane, unable to shuttle back and forth from the Golgi. As a result of the disrupted activity of the ATP7A protein, copper is poorly distributed to cells in the body. Copper accumulates in some tissues, such as the small intestine and kidneys, while the brain and other tissues have unusually low levels.[8][9] The decreased supply of copper can reduce the activity of numerous copper-containing enzymes that are necessary for the structure and function of bone, skin, hair, blood vessels, and the nervous system.[8][10] Copper is also critical for the propagation of prion proteins, and mice with mutations in Atp7a have a delayed onset of prion disease. [15] A comprehensive resource of clinically annotated genetic variants in ATP7A gene has been made available[16] confirming to the American College of Medical Genetics and Genomics guidelines for interpretation of sequence variants.
Inhibition
A proton pump inhibitor, Omeprazole, has been shown to block ATP7A, in addition to its more established role of blocking ATP4A.
See also
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000165240 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000033792 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ Tümer Z, Møller LB, Horn N (1999). "Mutation Spectrum of ATP7A, the Gene Defective in Menkes Disease". Copper Transport and Its Disorders. Advances in Experimental Medicine and Biology. Vol. 448. pp. 83–95. doi:10.1007/978-1-4615-4859-1_7. ISBN 978-1-4613-7204-2. PMID 10079817.
- 1 2 Kodama H, Murata Y (Aug 1999). "Molecular genetics and pathophysiology of Menkes disease". Pediatrics International. 41 (4): 430–5. doi:10.1046/j.1442-200x.1999.01091.x. PMID 10453200. S2CID 19509148.
- ↑ Šimčíková D, Heneberg P (December 2019). "Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases". Scientific Reports. 9 (1): 18577. Bibcode:2019NatSR...918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
- 1 2 3 4 5 Lutsenko S, Gupta A, Burkhead JL, Zuzel V (Aug 2008). "Cellular multitasking: the dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance". Archives of Biochemistry and Biophysics. 476 (1): 22–32. doi:10.1016/j.abb.2008.05.005. PMC 2556376. PMID 18534184.
- 1 2 3 4 5 Bertini I, Gray H, Stiefel E, Valentine J (2006). Biological inorganic chemistry : structure and reactivity. Sausalito, CA: University Science Books. ISBN 978-1-891389-43-6.
- ↑ Inesi G, Pilankatta R, Tadini-Buoninsegni F (Oct 2014). "Biochemical characterization of P-type copper ATPases". The Biochemical Journal. 463 (2): 167–76. doi:10.1042/BJ20140741. PMC 4179477. PMID 25242165.
- ↑ Banci L, Bertini I, Cantini F, Ciofi-Baffoni S (Aug 2010). "Cellular copper distribution: a mechanistic systems biology approach". Cellular and Molecular Life Sciences. 67 (15): 2563–89. doi:10.1007/s00018-010-0330-x. PMID 20333435. S2CID 41967295.
- ↑ Hordyjewska A, Popiołek Ł, Kocot J (August 2014). "The many "faces" of copper in medicine and treatment". Biometals. 27 (4): 611–21. doi:10.1007/s10534-014-9736-5. PMC 4113679. PMID 24748564.
- ↑ Siggs OM, Cruite JT, Du X, Rutschmann S, Masliah E, Beutler B, Oldstone MB (August 2012). "Disruption of copper homeostasis due to a mutation of Atp7a delays the onset of prion disease". Proc. Natl. Acad. Sci. U.S.A. 109 (34): 13733–8. doi:10.1073/pnas.1211499109. PMC 3427069. PMID 22869751.
- ↑ "ATP7Agen a comprehensive resource for clinically annotated variants in ATP7A Gene". clingen.igib.res.in. Retrieved 2020-07-06.
Further reading
- Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S (2005). "The copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum". J Biol Chem. 280 (10): 9640–5. doi:10.1074/jbc.M413840200. PMID 15634671.
- Greenough M, Pase L, Voskoboinik I, Petris MJ, O'Brien AW, Camakaris J (2004). "Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells". Am J Physiol Cell Physiol. 287 (5): C1463–71. doi:10.1152/ajpcell.00179.2004. PMID 15269005. S2CID 40168524.
- Møller LB, Tümer Z, Lund C, Petersen C, Cole T, Hanusch R, Seidel J, Jensen LR, Horn N (2000). "Similar splice-site mutations of the ATP7A gene lead to different phenotypes: classical Menkes disease or occipital horn syndrome". Am J Hum Genet. 66 (4): 1211–20. doi:10.1086/302857. PMC 1288188. PMID 10739752.
- Voskoboinik I, Camakaris J (2002). "Menkes copper-translocating P-type ATPase (ATP7A): biochemical and cell biology properties, and role in Menkes disease". J Bioenerg Biomembr. 34 (5): 363–71. doi:10.1023/A:1021250003104. PMID 12539963. S2CID 23109512.
- Harris ED, Reddy MC, Qian Y, Tiffany-Castiglioni E, Majumdar S, Nelson J (1999). "Multiple Forms of the Menkes Cu-ATPase". Copper Transport and Its Disorders. Advances in Experimental Medicine and Biology. Vol. 448. pp. 39–51. doi:10.1007/978-1-4615-4859-1_4. ISBN 978-1-4613-7204-2. PMID 10079814.
- Cox DW, Moore SD (2003). "Copper transporting P-type ATPases and human disease". J. Bioenerg. Biomembr. 34 (5): 333–8. doi:10.1023/A:1021293818125. PMID 12539960. S2CID 21471699.
- Voskoboinik I, Camakaris J (2003). "Menkes copper-translocating P-type ATPase (ATP7A): biochemical and cell biology properties, and role in Menkes disease". J. Bioenerg. Biomembr. 34 (5): 363–71. doi:10.1023/A:1021250003104. PMID 12539963. S2CID 23109512.
- La Fontaine S, Mercer JF (2007). "Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis". Arch. Biochem. Biophys. 463 (2): 149–67. doi:10.1016/j.abb.2007.04.021. PMID 17531189.
- Lutsenko S, LeShane ES, Shinde U (2007). "Biochemical basis of regulation of human copper-transporting ATPases". Arch. Biochem. Biophys. 463 (2): 134–48. doi:10.1016/j.abb.2007.04.013. PMC 2025638. PMID 17562324.
- Dierick HA, Ambrosini L, Spencer J, Glover TW, Mercer JF (1996). "Molecular structure of the Menkes disease gene (ATP7A)". Genomics. 28 (3): 462–9. doi:10.1006/geno.1995.1175. PMID 7490081.
- Tümer Z, Vural B, Tønnesen T, Chelly J, Monaco AP, Horn N (1995). "Characterization of the exon structure of the Menkes disease gene using vectorette PCR". Genomics. 26 (3): 437–42. doi:10.1016/0888-7543(95)80160-N. PMID 7607665.
- Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA (1995). "Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus". Nat. Genet. 8 (2): 195–202. doi:10.1038/ng1094-195. PMID 7842019. S2CID 12122103.
- Das S, Levinson B, Whitney S, Vulpe C, Packman S, Gitschier J (1994). "Diverse mutations in patients with Menkes disease often lead to exon skipping". Am. J. Hum. Genet. 55 (5): 883–9. PMC 1918324. PMID 7977350.
- Chelly J, Tümer Z, Tønnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP (1993). "Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein". Nat. Genet. 3 (1): 14–9. doi:10.1038/ng0193-14. PMID 8490646. S2CID 205341350.
- Mercer JF, Livingston J, Hall B, Paynter JA, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D (1993). "Isolation of a partial candidate gene for Menkes disease by positional cloning". Nat. Genet. 3 (1): 20–5. doi:10.1038/ng0193-20. PMID 8490647. S2CID 9148871.
- Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J (1993). "Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase". Nat. Genet. 3 (1): 7–13. doi:10.1038/ng0193-7. PMID 8490659. S2CID 24883244.
- Levinson B, Conant R, Schnur R, Das S, Packman S, Gitschier J (1997). "A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome". Hum. Mol. Genet. 5 (11): 1737–42. doi:10.1093/hmg/5.11.1737. PMID 8923001.
- Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD (1997). "Biochemical characterization and intracellular localization of the Menkes disease protein". Proc. Natl. Acad. Sci. U.S.A. 93 (24): 14030–5. doi:10.1073/pnas.93.24.14030. PMC 19489. PMID 8943055.
- Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1997). "Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking". EMBO J. 15 (22): 6084–95. doi:10.1002/j.1460-2075.1996.tb00997.x. PMC 452430. PMID 8947031.
- Tümer Z, Lund C, Tolshave J, Vural B, Tønnesen T, Horn N (1997). "Identification of point mutations in 41 unrelated patients affected with Menkes disease". Am. J. Hum. Genet. 60 (1): 63–71. PMC 1712537. PMID 8981948.
- Dierick HA, Adam AN, Escara-Wilke JF, Glover TW (1997). "Immunocytochemical localization of the Menkes copper transport protein (ATP7A) to the trans-Golgi network". Hum. Mol. Genet. 6 (3): 409–16. doi:10.1093/hmg/6.3.409. PMC 7185191. PMID 9147644.
- Ronce N, Moizard MP, Robb L, Toutain A, Villard L, Moraine C (1997). "A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family". Am. J. Hum. Genet. 61 (1): 233–8. doi:10.1016/S0002-9297(07)64297-9. PMC 1715861. PMID 9246006.
- Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ (1998). "Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATPase". Nat. Struct. Biol. 5 (1): 47–54. doi:10.1038/nsb0198-47. PMID 9437429. S2CID 172550.
External links
- ATP7A+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- GeneReviews/NCBI/NIH/UW entry on ATP7A-Related Copper Transport Disorders Includes: Menkes Disease, Occipital Horn Syndrome, ATP7A-Related Distal Motor Neuropathy
- OMIM entries on ATP7A-Related Copper Transport Disorders
- GeneCard
- Human ATP7A genome location and ATP7A gene details page in the UCSC Genome Browser.
PDB gallery | |
|---|---|
|