| Abetalipoproteinemia | |
|---|---|
| Other names | Bassen-Kornzweig syndrome[1] |
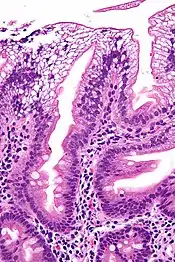 | |
| Micrograph showing enterocytes with a clear cytoplasm (due to lipid accumulation) characteristic of abetalipoproteinemia. Duodenal biopsy. H&E stain. | |
| Specialty | Endocrinology |
Abetalipoproteinemia (also known as: Bassen–Kornzweig syndrome, microsomal triglyceride transfer protein deficiency disease, MTP deficiency, and betalipoprotein deficiency syndrome[2]) is a disorder characterized by abnormal absorption of fat and fat-soluble vitamins from food.[3] It is caused by a mutation in microsomal triglyceride transfer protein resulting in deficiencies in the apolipoproteins B-48 and B-100, which are used in the synthesis and exportation of chylomicrons and VLDL respectively. It is not to be confused with familial dysbetalipoproteinemia.
It is a rare autosomal recessive disorder.[4]
Presentation
Symptoms
Often symptoms will arise that indicate the body is not absorbing or making the lipoproteins that it needs. These symptoms usually appear en masse.
These symptoms come as follows:
- Failure to thrive (i.e. failure to grow in infancy)[5]
- Steatorrhea (i.e. fatty, pale stools)[5][6]
- Frothy stools[5]
- Foul smelling stools[5]
- Protruding abdomen
- Intellectual disability/developmental delay
- Developmental coordination disorder, evident by age ten
- Ataxia
- Muscle weakness
- Slurred speech (dysarthria)[7]
- Scoliosis (curvature of the spine)
- Progressive decreased vision
- Balance and coordination problems
Signs
Features
Abetalipoproteinemia affects the absorption of dietary fats, cholesterol, and certain vitamins. People affected by this disorder are not able to make certain lipoproteins, which are molecules that consist of proteins combined with cholesterol and particular fats called triglycerides. This leads to a multiple vitamin deficiency, affecting the fat-soluble vitamin A, vitamin D, vitamin E, and vitamin K.[9] However, many of the observed effects are due to vitamin E deficiency in particular.[9]

Signs and symptoms vary and present differently from person to person. In general, 80–99% of individuals exhibit malabsorption of fats and fat-soluble vitamins. Approximately 30–79% of people with the disease display symptoms related to abnormality of the retinal pigmentation, ataxia, muscular hypotonia or reduced tendon reflexes.[2]
The signs and symptoms of abetalipoproteinemia appear in the first few months of life (because pancreatic lipase is not active in this period). They can include failure to gain weight and grow at the expected rate (failure to thrive); diarrhea; abnormal spiny red blood cells (acanthocytosis); and fatty, foul-smelling stools (steatorrhea).[9] The stool may contain large chunks of fat and/or blood. Infants often present with gastrointestinal problems caused by the poor fat absorption, which also contributes to steatorrhea. Other features of this disorder may develop later in childhood and often impair the function of the nervous system. They can include poor muscle coordination, difficulty with balance and movement (ataxia),[9][10] and progressive degeneration of the retina (the light-sensitive layer in the posterior eye) that can progress to near-blindness (due to deficiency of vitamin A, retinol).[9] Adults in their thirties or forties may have increasing difficulty with balance and walking. Many of the signs and symptoms of abetalipoproteinemia result from a severe vitamin deficiency, especially vitamin E deficiency, which typically results in eye problems with degeneration of the spinocerebellar and dorsal column tracts.
Genetics
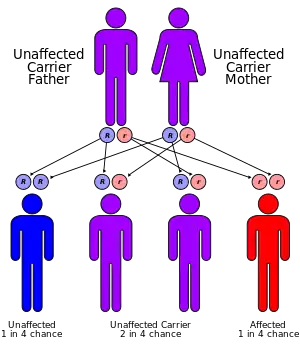
Mutations in the microsomal triglyceride transfer protein gene (MTTP) have been associated with this condition[9] (apolipoprotein B deficiency, a related condition, is associated with deficiencies of apolipoprotein B).[11]
The MTTP gene provides instructions for making a protein called microsomal triglyceride transfer protein, which is essential for creating beta-lipoproteins.[12] These lipoproteins are both necessary for the absorption of fats, cholesterol, and fat-soluble vitamins from the diet and necessary for the efficient transport of these substances in the bloodstream.[13] Most of the mutations in this gene lead to the production of an abnormally short microsomal triglyceride transfer protein, which prevents the normal creation of beta-lipoproteins in the body.[14] MTTP-associated mutations are inherited in an autosomal recessive pattern, which means both copies of the gene must be faulty to produce the disease.[14]
The disease is extremely rare with approximately 100 reported cases worldwide since it was first identified by doctors Bassen and Kornzweig in 1950.[3]
Mechanism
Abetalipoproteinemia effects multiple physiological systems, the two most common being the nervous and the skeletal. Disruption of nervous function includes loss of reflexes, speech impairments, tremors or involuntary motor tics, or peripheral neuropathy (damage to the nerves outside of the brain and spinal cord). Peripheral neuropathy causes loss of sensation, weakness or numbness and pain in the extremities through stabbing, burning, or tingling sensations.[15] Skeletal system developments often include lordosis, kyphoscoliosis, or pes cavus.[3] Individuals often have abnormal bleeding due to the difficulty of forming clots.
Additional complications of the diseases if not properly treated include blindness, mental deterioration, ataxia, loss of peripheral nerve function.
Diagnosis
The initial workup of Abetalipoproteinemia typically consists of stool sampling, a blood smear, and a fasting lipid panel, though these tests are not confirmatory.[16] As the disease is rare, though a genetics test is necessary for diagnosis, it is generally not done initially. However, prenatal testing may be available for pregnancies identified to be at an increased risk (if both parents are unaffected carrier or one parent is affected and the other in a carrier).
Acanthocytes are seen on blood smear.[17] Since there is no or little assimilation of chylomicrons, their levels in plasma remains low.
The inability to absorb fat in the ileum will result in steatorrhea, or fat in the stool. As a result, this can be clinically diagnosed when foul-smelling stool is encountered. Low levels of plasma chylomicron are also characteristic.
There is an absence of apolipoprotein B. On intestinal biopsy, vacuoles containing lipids are seen in enterocytes. This disorder may also result in fat accumulation in the liver (hepatic steatosis). Because the epithelial cells of the bowel lack the ability to place fats into chylomicrons, lipids accumulate at the surface of the cell, crowding the functions that are necessary for proper absorption.
Multiple related disorders present with similar symptoms as abetalipoproteinemia that can provide a useful diagnosis through comparisons. Some of those disorders are:
Treatment
Treatment normally consists of rigorous dieting, involving massive amounts of vitamin E.[10] High-dose Vitamin E therapy helps the body restore and produce lipoproteins, which people with abetalipoproteinemia usually lack. Vitamin E also helps keep skin and eyes healthy; studies show that many affected males will have vision problems later on in life. Common additional supplementation includes medium chain fatty acids and linoleic acid. Treatments also aim to slow the progression of nervous system abnormalities. Developmental coordination disorder and muscle weakness are usually treated with physiotherapy or occupational therapy. Dietary restriction of triglycerides has also been useful. Nutritionists often work with medical professionals to design appropriate dietary treatments for their patients.[2]
Prognosis
Prognosis can vary heavily based on the severity of the neurological dysfunction. If treatment is initiated early in disease the neurologic sequelae may be reversed and further deterioration can be prevented.[18] Long-term outlook is reasonably good for most people when diagnosed and treated early. A case study presented a female patient diagnosed at the age of 11. Despite the relatively late diagnosis, the patient married and at the age of 34, gave birth to a full-term healthy infant. Her medication included vitamin K 10 mg twice a week, beta-carotene 40,000 IU daily, vitamin A 10,000 IU daily, vitamin E 400 IU daily, vitamins B6 and B12, calcium, magnesium and eye drops.[19]
Prolonged vitamin deficiencies can further compromise health. Specifically, a prolonged vitamin E deficiency can lead to the development of limiting ataxia and gait disturbances. Some individuals may develop retinal degeneration and blindness. If left untreated, the condition may lead to death.[20]
Current research
A primary goal of abetalipoproteinemia research is to supply the fat-soluble vitamins the body lacks in the disease. Previous research considered the short-term use of intravenous infusion of vitamins A and E. The goal was to determine whether these infusions would delay or counteract the symptoms in patients. No results were posted.[21]
More recent research has focused on different ways to supply the patient with vitamin E. In 2018, the Journal of Lipid Research published a study testing alternative forms of vitamin E absorption. Currently, vitamin E is most often supplemented in the fat-soluble form vitamin E acetate. Due to fat malabsorption, the intended supplementation is considerably compromised. Two different forms were tested: vitamin E tocofersolan and α-tocopherol acetate. The study concluded that plasma bioavailabilities were extremely low (2.8% and 3.1%, respectively). Additionally, plasma concentrations of tocopherol were not significantly different in patients.[22]
This study provides new insight in vitamin E supplementation and suggests further research is needed with different forms of vitamin E as possible treatment options to abetalipoproteinemia.
Currently, there is a clinical study recruiting abetalipoproteinemia patients to study inherited retinal degenerative disease.[23]
References
- ↑ Bassen FA, Kornzweig AL (April 1950). "Malformation of the erythrocytes in a case of atypical retinitis pigmentosa". Blood. 5 (4): 381–87. doi:10.1182/blood.V5.4.381.381. PMID 15411425.
- 1 2 3 "Abetalipoproteinemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2019-10-14. Retrieved 2019-11-06.
- 1 2 3 "Abetalipoproteinemia". Genetics Home Reference. Retrieved 2018-04-18.
- ↑ Benayoun L, Granot E, Rizel L, Allon-Shalev S, Behar DM, Ben-Yosef T (April 2007). "Abetalipoproteinemia in Israel: evidence for a founder mutation in the Ashkenazi Jewish population and a contiguous gene deletion in an Arab patient". Molecular Genetics and Metabolism. 90 (4): 453–7. doi:10.1016/j.ymgme.2006.12.010. PMID 17275380.
- 1 2 3 4 5 Hasosah MY, Shesha SJ, Sukkar GA, Bassuni WY (October 2010). "Rickets and dysmorphic findings in a child with abetalipoproteinemia". Saudi Medical Journal. 31 (10): 1169–71. PMID 20953537.
- 1 2 Moutzouri E, Elisaf M, Liberopoulos EN (March 2011). "Hypocholesterolemia". Current Vascular Pharmacology. 9 (2): 200–12. doi:10.2174/157016111794519354. PMID 20626336.
- ↑ Genetics Home Reference. (n.d.). Abetalipoproteinemia. In MedlinePlus Genetics. Retrieved June 26, 2021, from https://medlineplus.gov/genetics/condition/abetalipoproteinemia/
- ↑ Cooper RA, Durocher JR, Leslie MH (July 1977). "Decreased fluidity of red cell membrane lipids in abetalipoproteinemia". The Journal of Clinical Investigation. 60 (1): 115–21. doi:10.1172/JCI108747. PMC 372349. PMID 874076.
- 1 2 3 4 5 6 "Abetalipoproteinemia - Genetics Home Reference". Retrieved 2008-02-24.
- 1 2 Hentati F, El-Euch G, Bouhlal Y, Amouri R (2012). "Ataxia with vitamin E deficiency and abetalipoproteinemia". Ataxic Disorders. Handbook of Clinical Neurology. Vol. 103. pp. 295–305. doi:10.1016/B978-0-444-51892-7.00018-8. ISBN 9780444518927. PMID 21827896.
- ↑ Hussain MM, Rava P, Walsh M, Rana M, Iqbal J (February 2012). "Multiple functions of microsomal triglyceride transfer protein". Nutrition & Metabolism. 9: 14. doi:10.1186/1743-7075-9-14. PMC 3337244. PMID 22353470.
- ↑ Najah M, Youssef SM, Yahia HM, Afef S, Awatef J, Saber H, et al. (April 2013). "Molecular characterization of Tunisian families with abetalipoproteinemia and identification of a novel mutation in MTTP gene". Diagnostic Pathology. 8 (1): 54. doi:10.1186/1746-1596-8-54. PMC 3632489. PMID 23556456.
- ↑ Magnolo L, Najah M, Fancello T, Di Leo E, Pinotti E, Brini I, et al. (January 2013). "Novel mutations in SAR1B and MTTP genes in Tunisian children with chylomicron retention disease and abetalipoproteinemia". Gene. 512 (1): 28–34. doi:10.1016/j.gene.2012.09.117. PMID 23043934.
- 1 2 Pons V, Rolland C, Nauze M, Danjoux M, Gaibelet G, Durandy A, et al. (July 2011). "A severe form of abetalipoproteinemia caused by new splicing mutations of microsomal triglyceride transfer protein (MTTP)". Human Mutation. 32 (7): 751–9. doi:10.1002/humu.21494. PMID 21394827. S2CID 28693034.
- ↑ "Peripheral neuropathy - Symptoms and causes". Mayo Clinic. Retrieved 2019-11-06.
- ↑ Demircioğlu F, Oren H, Yilmaz S, Arslan N, Gürcü O, Irken G (August 2005). "Abetalipoproteinemia: importance of the peripheral blood smear". Pediatric Blood & Cancer. 45 (2): 237. doi:10.1002/pbc.20360. PMID 15765527. S2CID 28481550.
- ↑ Ozsoylu S (Jan–Feb 2011). "Red cells in abetalipoproteinemia". The Turkish Journal of Pediatrics. 53 (1): 119. PMID 21534356.
- ↑ Rader DJ, Brewer HB (August 1993). "Abetalipoproteinemia. New insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease". JAMA. 270 (7): 865–9. doi:10.1001/jama.1993.03510070087042. PMID 8340987.
- ↑ Zamel R, Khan R, Pollex RL, Hegele RA (July 2008). "Abetalipoproteinemia: two case reports and literature review". Orphanet Journal of Rare Diseases. 3 (1): 19. doi:10.1186/1750-1172-3-19. PMC 2467409. PMID 18611256.
- ↑ "Abetalipoproteinemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2019-10-14. Retrieved 2018-04-18.
- ↑ "Vitamin Replacement in Abetalipoproteinemia - Full Text View - ClinicalTrials.gov". clinicaltrials.gov. 3 March 2008. Retrieved 2019-12-13.
- ↑ Cuerq C, Henin E, Restier L, Blond E, Drai J, Marçais C, et al. (September 2018). "Efficacy of two vitamin E formulations in patients with abetalipoproteinemia and chylomicron retention disease". Journal of Lipid Research. 59 (9): 1640–1648. doi:10.1194/jlr.M085043. PMC 6121919. PMID 30021760.
- ↑ Clinical trial number NCT02435940 for "Inherited Retinal Degenerative Disease Registry" at ClinicalTrials.gov
External links
- Abetalipoproteinemia at NLM Genetics Home Reference