Aluminium(I) nucleophiles are a group of inorganic and organometallic nucleophilic compounds containing at least one aluminium metal center in the +1 oxidation state with a lone pair of electrons strongly localized on the aluminium(I) center.
Prevalent aluminium(III) compounds such as aluminium trihalides (AlCl3, AlBr3, AlI3) are regularly employed in organic synthesis as electrophiles or Lewis acids. However, upon reducing of the metal center, aluminium(I) compounds may gain a lone pair which confers them nucleophilic character. While many aluminium(I) compounds are thermodynamically unstable due to their low oxidation state and act as good reducing agents, recent synthetic developments allowed for the isolation of stable aluminium(I) compounds. The first example of an isolable aluminium(I) compound was the tetrameric (AlCp*)4 (Cp* = pentamethylcyclopentadienyl) reported by Schnöckel and coworkers in 1991,[1] while the first monomeric aluminium(I) compound was isolated on a β-diketiminate NacNac-type ligand by Roesky and coworkers in 2000.[2] This initial monomeric aluminium(I) neutral compound and other closely related β-diketiminate supported aluminium(I) compounds were predicted to display ambiphilic behavior: electrophilic character due to the formally vacant aluminium p-orbital, as well as nucleophilic character due to the presence of a lone pair. However, in practice, their nucleophilic character was not observed through coordination to electrophiles or nucleophilic substitution; instead, their main mode of reactivity involves oxidative addition pathways, due to the low oxidation state of the aluminium center.[3]
Nevertheless, these seminal findings along with more recent advances in the synthesis, isolation, and characterization of aluminium(I) compounds allowed for the discovery of a novel type of reactivity at aluminium centers: nucleophilicity at aluminium(I) anions, referred to as aluminyl anions.[4] The first isolation of a nucleophilic aluminium center was achieved in 2018 by Aldridge, Goicoechea and coworkers when they were able to synthesize the first aluminyl anion,[5] following the discoveries of gallium[6][7][8] and indium[9] analogues, heavier group 13 analogues which are more stable than aluminium in the lower +1 oxidation state. Since then, several other nucleophilic aluminyl anions have been synthesized and characterized.
Synthesis and Structure
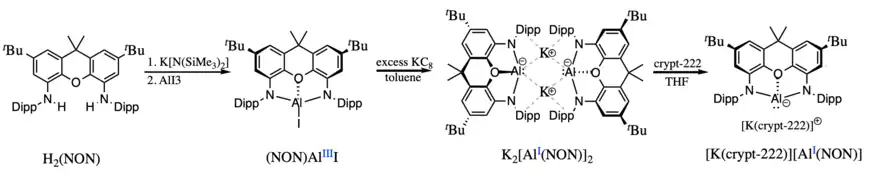
The most common ligands employed so far in the synthesis of nucleophilic aluminyl anions are diamido dianions. The first aluminyl anion to be isolated by Aldridge, Goicoechea and coworkers is supported by a xanthene-based diamido ligand, referred to as NON by the authors. In its synthesis, the xanthene-dianiline H2NON was first deprotonated in the presence of a strong base. Metallation of the NON ligand proceeded via aluminium triodide addition, forming an aluminium(III) metal center. The aluminium(III) center was then reduced via addition of an excess of a strong reducing agent, potassium graphite (KC8), to the aluminyl species. Following the two electron reduction, a bright-yellow dimeric species with the chemical formula K2[Al(NON)]2 was formed, as confirmed by X-ray crystallography.[5] The compound was found to be stable for several days at room temperature, both in the solid state and in a benzene solution. Each aluminium(I) atom is supported by two amido nitrogen atoms and one ethereal oxygen atom of the ligand, formally having a filled octet. The two monomeric units are joined by electrostatic interactions between the positively charged potassium cations and the electron clouds of the Dipp (diisopropylphenyl) substituents. The crystal structure confirms no aluminium-aluminium interactions, while the dimeric structure is preserved in solution, as highlighted by diffusion ordered spectroscopy (DOSY) NMR experiments.[5] However, in the presence of chelating agent [2.2.2]-cryptand (crypt-222), the potassium cation could be encapsulated, leading to a charge-separated complex with a "naked" aluminyl anion, the first such anion to be reported.[10] Following this initial synthesis, other nucleophilic diamido aluminyl anions have been reported, also synthesized through the reduction of the corresponding aluminium(III) iodide with excess potassium graphite and also isolated as a dimeric species with a similar structure to that of K2[Al(NON)]2.[11][12]
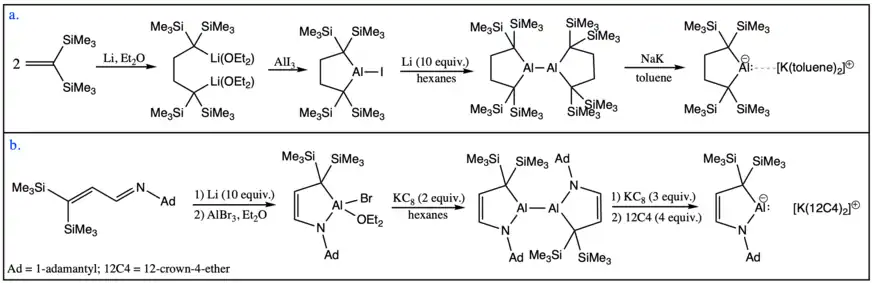
Other nucleophilic aluminyl anions include alkyl substituents on the metal center. The first aluminyl anion of this type was reported by Yamashita and coworkers in 2020. The dialkylaluminium(III) iodide serves as a key intermediate in its synthesis as well but, in this case, the reduction occurred in two subsequent steps generating an isolable dialumane intermediate. Following the second reduction step of the dialumane, the dialkyl aluminyl anion was isolated as a monomer stabilized through weak electrostatic interactions with the potassium based cation. In this case, encapsulation of the potassium cation to generate a charge-separated ionic pair was not possible.[13] The second example of an alkyl aluminyl system was synthesized by Kinjo and coworkers in 2020 also through the reduction of a dialumane intermediate to form the second charge-separated aluminyl anion.[14]
Electronic Structure
_nucleophile.png.webp)
Initial electronic structure calculations of aluminium(I) nucleophiles were gained using ab initio calculations. Cimpoesu and coworkers analyzed the first monomeric aluminium(I) compound by plotting the contour map of the Laplacian of the electron density inside the molecule and found that there is indeed a lone pair localized on the aluminium atom, in a spherical geometry, pointing outside of the ring.[15] Their computational analysis confirmed that the aluminium lone pair is localized in an s orbital, in good agreement with the aluminium(I) atom having an s2 electron configuration.
More recent computational electronic structure analyses of isolated aluminyl anions demonstrate that the trends in their electronic structure follows those of singlet carbenes, which also have a high-lying filled orbital (a lone pair) and a low-lying empty p orbital on the same atom.[4] In all cases, the highest occupied molecular orbital (HOMO) of aluminyl anions is the aluminium lone pair which confers these compounds nucleophilic character. Unsurprisingly, the diamido aluminyl compounds have lower energy HOMOs than alkyl-amido or dialkyl aluminyls, owing to the presence of adjacent electronegative nitrogen atoms. Moreover, adjacent nitrogen atoms can π-donate into the empty p orbitals of the aluminium(I) centers, increasing the energy of this empty orbital, as verified computationally. In fact, the aluminium empty p orbital has a significant contribution to the aluminyl lowest unoccupied molecular orbital (LUMO) only in the case of dialkyl aluminyls. For mono- or diamido aluminyls, the aluminium empty p orbital is higher in energy than the ligand-based LUMO. Nevertheless, all analyzed aluminyl systems have a pretty similar aluminium lone pair - aluminium empty p orbital energetic gap of 3.42 to 4.06 eV, while the dialkyl aluminyl systems were, as expected, found to have the lowest HOMO-LUMO gap.[4]
Reactivity
Aluminyl anions can engage in a variety of reactions, including oxidative addition reactions with strong single bonds such as H-H, C-H, N-H, or Si-H bonds,[5][10][13][18][19] and oxidation reactions in the presence of oxidizing agents,[18][20][21][22] both characteristics of the low oxidation state of the aluminium(I) center. In addition, the aluminyl anions have been shown to undergo cycloaddition reactions with alkenes, alkynes, and polyaromatics, similar to singlet carbenes, which can be attributed to the ambiphilic character of the aluminium(I) center.[10][11][23] However, their most striking reactivity mode is that as nucleophiles.
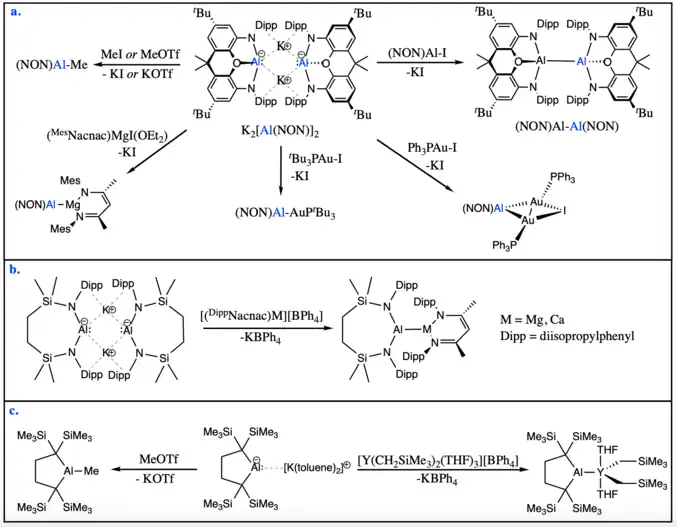
While all aluminyl anions display nucleophilic character, a few of them have been shown to participate in nucleophilic substitution reactions with carbon-based or metal-based electrophiles. Aldridge and Goicoechea's diamido aluminyl anion, K2[Al(NON)]2, can react with methyl electrophiles such as methyl iodide and methyl triflate to yield the corresponding methyl-aluminium product. The same aluminyl system can react with bulky metal iodide electrophiles in nucleophilic substitution reactions to form new aluminium-metal bonds, including aluminium-aluminium, aluminium-magnesium, or aluminium-gold bonds.[5][16]
Similarly, Hill and McMullin's dimeric diamido aluminyl anion can react with magnesium or calcium electrophiles to form new metal-metal bonds.[12]
Additionally, Yamashita's dialkyl aluminyl anion can also act as a nucleophile in nucleophilic substitution reactions with electrophiles such as methyl triflate or metal centers. Using this method, the formation of an aluminium-yttrium bond was achieved.[13][17]
It has been noted that in all recorded nucleophilic substitutions using aluminyl nucleophiles have been done in the presence of weakly coordinating leaving groups or anions (iodide, triflate, or tetraphenylborate) to avoid coordination of the anion to the metal centers.[4]
See also
References
- ↑ Dohmeier, Carsten; Robl, Christian; Tacke, Matthias; Schnöckel, Hansgeorg (1991). "The Tetrameric Aluminum(I) Compound [Al(η5-C5Me5)4]". Angewandte Chemie International Edition in English. 30 (5): 564–565. doi:10.1002/anie.199105641. ISSN 1521-3773.
- ↑ Cui, Chunming; Roesky, Herbert W.; Schmidt, Hans-Georg; Noltemeyer, Mathias; Hao, Haijun; Cimpoesu, Fanica (2000). "Synthesis and Structure of a Monomeric Aluminum(I) Compound [HC(CMeNAr)2Al] (Ar=2,6–iPr2C6H3): A Stable Aluminum Analogue of a Carbene". Angewandte Chemie International Edition. 39 (23): 4274–4276. doi:10.1002/1521-3773(20001201)39:23<4274::AID-ANIE4274>3.0.CO;2-K. ISSN 1521-3773. PMID 29711904.
- ↑ Zhong, Mingdong; Sinhababu, Soumen; Roesky, Herbert W. (2020-02-05). "The unique β-diketiminate ligand in aluminum(I) and gallium(I) chemistry". Dalton Transactions. 49 (5): 1351–1364. doi:10.1039/C9DT04763H. ISSN 1477-9234. PMID 31942579.
- 1 2 3 4 Hicks, Jamie; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (2020-08-18). "The Aluminyl Anion: A New Generation of Aluminium Nucleophile". Angewandte Chemie International Edition. 60 (4): 1702–1713. doi:10.1002/anie.202007530. ISSN 1433-7851. PMID 32567755. S2CID 219973175.
- 1 2 3 4 5 6 7 Hicks, Jamie; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (May 2018). "Synthesis, structure and reaction chemistry of a nucleophilic aluminyl anion". Nature. 557 (7703): 92–95. doi:10.1038/s41586-018-0037-y. ISSN 0028-0836. PMID 29662211. S2CID 51968151.
- ↑ Schmidt, Eva S.; Jockisch, Alexander; Schmidbaur, Hubert (1999-10-01). "A Carbene Analogue with Low-Valent Gallium as a Heteroatom in a quasi-Aromatic Imidazolate Anion". Journal of the American Chemical Society. 121 (41): 9758–9759. doi:10.1021/ja9928780. ISSN 0002-7863.
- ↑ Baker, Robert J.; Farley, Robert D.; Jones, Cameron; Kloth, Marc; Murphy, Damien M. (2002-10-03). "The reactivity of diazabutadienes toward low oxidation state Group 13 iodides and the synthesis of a new gallium(I) carbene analogue". Journal of the Chemical Society, Dalton Transactions (20): 3844–3850. doi:10.1039/B206605J. ISSN 1364-5447.
- ↑ Fedushkin, Igor L.; Lukoyanov, Anton N.; Fukin, Georgy K.; Ketkov, Sergey Yu.; Hummert, Markus; Schumann, Herbert (2008-09-26). "Synthesis, Molecular Structure and DFT Study of [(dpp-bian)Ga—M(Et2O)3] (M=Li, Na; dpp-bian=1,2-bis[(2,6-diisopropylphenyl)imino]acenaphthene)". Chemistry – A European Journal. 14 (28): 8465–8468. doi:10.1002/chem.200801267. ISSN 0947-6539. PMID 18698564.
- ↑ Schwamm, Ryan J.; Anker, Mathew D.; Lein, Matthias; Coles, Martyn P.; Fitchett, Christopher M. (2018-05-14). "Indyllithium and the Indyl Anion [InL] − : Heavy Analogues of N-Heterocyclic Carbenes". Angewandte Chemie International Edition. 57 (20): 5885–5887. doi:10.1002/anie.201802444. PMID 29575533.
- 1 2 3 Hicks, Jamie; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (2019-07-17). "Reversible, Room-Temperature C—C Bond Activation of Benzene by an Isolable Metal Complex". Journal of the American Chemical Society. 141 (28): 11000–11003. doi:10.1021/jacs.9b05925. ISSN 0002-7863. PMID 31251586. S2CID 195760502.
- 1 2 Schwamm, Ryan J.; Anker, Mathew D.; Lein, Matthias; Coles, Martyn P. (2019). "Reduction vs. Addition: The Reaction of an Aluminyl Anion with 1,3,5,7-Cyclooctatetraene". Angewandte Chemie International Edition. 58 (5): 1489–1493. doi:10.1002/anie.201811675. ISSN 1521-3773. PMID 30548141. S2CID 56484091.
- 1 2 3 Schwamm, Ryan J.; Coles, Martyn P.; Hill, Michael S.; Mahon, Mary F.; McMullin, Claire L.; Rajabi, Nasir A.; Wilson, Andrew S. S. (2020-03-02). "A Stable Calcium Alumanyl". Angewandte Chemie International Edition. 59 (10): 3928–3932. doi:10.1002/anie.201914986. ISSN 1433-7851. PMC 7159655. PMID 31830364.
- 1 2 3 4 5 Kurumada, Satoshi; Takamori, Shuhei; Yamashita, Makoto (January 2020). "An alkyl-substituted aluminium anion with strong basicity and nucleophilicity". Nature Chemistry. 12 (1): 36–39. doi:10.1038/s41557-019-0365-z. ISSN 1755-4349. PMID 31767993. S2CID 208279573.
- 1 2 Koshino, Kota; Kinjo, Rei (2020-05-13). "Construction of σ-Aromatic AlB2 Ring via Borane Coupling with a Dicoordinate Cyclic (Alkyl)(Amino)Aluminyl Anion". Journal of the American Chemical Society. 142 (19): 9057–9062. doi:10.1021/jacs.0c03179. hdl:10356/151894. ISSN 0002-7863. PMID 32321239. S2CID 216084220.
- 1 2 Cui, Chunming; Roesky, Herbert W.; Schmidt, Hans-Georg; Noltemeyer, Mathias; Hao, Haijun; Cimpoesu, Fanica (2000). "Synthesis and Structure of a Monomeric Aluminum(I) Compound [HC(CMeNAr)2Al] (Ar=2,6–iPr2C6H3): A Stable Aluminum Analogue of a Carbene". Angewandte Chemie International Edition. 39 (23): 4274–4276. doi:10.1002/1521-3773(20001201)39:23<4274::AID-ANIE4274>3.0.CO;2-K. ISSN 1521-3773. PMID 29711904.
- 1 2 Hicks, Jamie; Mansikkamäki, Akseli; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (2019-01-21). "A nucleophilic gold complex". Nature Chemistry. 11 (3): 237–241. doi:10.1038/s41557-018-0198-1. ISSN 1755-4330. PMID 30664716. S2CID 58642643.
- 1 2 Sugita, Kengo; Yamashita, Makoto (2020-03-13). "An Alumanylyttrium Complex with an Absorption due to a Transition from the Al−Y Bond to an Unoccupied d‐Orbital". Chemistry – A European Journal. 26 (20): 4520–4523. doi:10.1002/chem.202000752. ISSN 0947-6539. PMID 32052882. S2CID 211101307.
- 1 2 Heilmann, Andreas; Hicks, Jamie; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (2020). "Carbon Monoxide Activation by a Molecular Aluminium Imide: C−O Bond Cleavage and C−C Bond Formation". Angewandte Chemie International Edition. 59 (12): 4897–4901. doi:10.1002/anie.201916073. ISSN 1521-3773. PMID 31999037. S2CID 210948048.
- ↑ Cabrera-Trujillo, Jorge Juan; Fernández, Israel (2020-09-10). "Rationalizing the AlI-Promoted Oxidative Addition of C−C Versus C−H Bonds in Arenes". Chemistry – A European Journal. 26 (51): 11806–11813. doi:10.1002/chem.202000921. ISSN 0947-6539. PMID 32329537. S2CID 216110827.
- ↑ Hicks, Jamie; Heilmann, Andreas; Vasko, Petra; Goicoechea, Jose M.; Aldridge, Simon (2019). "Trapping and Reactivity of a Molecular Aluminium Oxide Ion". Angewandte Chemie International Edition. 58 (48): 17265–17268. doi:10.1002/anie.201910509. ISSN 1521-3773. PMID 31550066. S2CID 202746925.
- ↑ Anker, Mathew D.; Coles, Martyn P. (2019). "Aluminium-Mediated Carbon Dioxide Reduction by an Isolated Monoalumoxane Anion". Angewandte Chemie International Edition. 58 (50): 18261–18265. doi:10.1002/anie.201911550. ISSN 1521-3773. PMID 31568609. S2CID 203624740.
- ↑ Anker, Mathew D.; Schwamm, Ryan J.; Coles, Martyn P. (2020-02-20). "Synthesis and reactivity of a terminal aluminium–imide bond". Chemical Communications. 56 (15): 2288–2291. doi:10.1039/C9CC09214E. ISSN 1364-548X. PMID 31984981. S2CID 210922664.
- ↑ Sugita, Kengo; Nakano, Ryo; Yamashita, Makoto (2020-02-17). "Cycloaddition of Dialkylalumanyl Anion toward Unsaturated Hydrocarbons in (1+2) and (1+4) Modes". Chemistry – A European Journal. 26 (10): 2174–2177. doi:10.1002/chem.201905830. ISSN 0947-6539. PMID 31880356. S2CID 209489996.