Autophagic vacuolar myopathy (AVM) consists of multiple rare genetic disorders with common histological and pathological features on muscle biopsy.[1] The features highlighted are vacuolar membranes of the autophagic vacuoles having sarcolemmal characteristics and an excess of autophagic vacuoles.[2] There are currently five types of AVM identified.[1] The signs and symptoms become more severe over the course of the disease. It begins with an inability to pick up small objects and progresses to difficulty in walking.[3] The age of onset varies from early childhood to late adulthood, affecting people of all ages.[4]
The disorders are caused by a mutation in different parts of the chromosome: Danon disease is caused by a mutation of the LAMP2 gene; XMEA is caused by mutations of the VMA21 gene.[5] These gene mutations slow down the fusion between autophagic vacuoles and lysosomes, leading to the accumulation of autophagic vacuoles.[5] The result is the breakdown of muscle cells, which attributes to muscle weakness in patients with AVM.[5] The mode of transmission is X-linked, with Danon Disease being X-linked dominant and XMEA being X-linked recessive.[6] Other types of AVM are less researched in terms of their mode of transmission, but it is known that these diseases are all gene-related.[6]
Diagnosis of AVM involves various types of genetic testing, alongside a thorough examination of the patient's history and symptoms.[1] Treatment of the disease currently involves Enzyme Replacement Therapy and gene therapy is a possibility for the future, a solution which may cure the disease completely.[7]
Categories and causes
There are five types of AVM: Danon disease, X-linked myopathy with excessive autophagy (XMEA), Pompe Disease, infantile AVM, and adult onset AVM with multiorgan involvement.[2]
Danon disease, XMEA and Pompe Disease are better researched in terms of the gene causing the disorder. XMEA is linked to mutations in the VMA21 gene at Xq28 while Danon disease and Pompe Disease are associated with LAMP2 gene located on the X chromosome and the GAA gene respectively.[5] For infantile AVM and adult onset AVM with multiorgan involvement, the LAMP2 gene is unrelated to the two diseases, though the specific genes related are unknown.[8]
The causes of the disease is different mutation occurring in the aforementioned genes. For Danon disease which is related to LAMP2, since the LAMP2 gene is responsible for the production of the LAMP-2 protein, which plays a role in the transport of cellular materials into the lysosome, mutations of the LAMP2 gene lead to little to no LAMP-2 protein production, impairing the transport of cellular materials into the lysosome.[5] Without the LAMP-2 protein, fusion between autophagic vacuoles and lysosomes occurs much slower, leading to the accumulation of autophagic vacuoles.[5] This accumulation leads to the breakdown of muscle cells, thereby causing the muscle weakness exhibited in Danon disease patients.[5]
Mode of inheritance
Most of the AVMs are sex-linked, meaning the gene with regards to AVM is located on a sex chromosome.[9] Danon disease is hereditary and is inherited in an X-linked dominant pattern.[10] The LAMP2 gene associated with Danon disease is located on the X-chromosome.[5] Since the X-chromosome is one of the two sex chromosomes, females may develop the disease with just one mutation in one of their two copies of the X-chromosome.[11] Since there is only one X-chromosome present in humans, a mutation in that particular X chromosome could already cause the disease.[11] Therefore, males typically exhibit more severe symptoms than females. Because Danon disease is X-linked, this means that fathers cannot pass their X-linked traits to their sons because males always pass their Y-chromosome to their male offsprings.[10]
Though also X-linked, the transmission pattern of XMEA is of a X-linked recessive pattern.[11] Since females have two X-chromosomes, this means that females may be carriers who are asymptomatic or only showing mild symptoms.[11] Meanwhile, males become diseased if they inherit just one X-chromosome that contains the mutated VMA21 gene.[11] Similar to Danon disease, males with XMEA cannot pass the mutated gene to their sons, but will pass the gene to their daughters, who will be carriers.[11] Female carriers have a subsequent 25% chance of having a carrier daughter, a 25% chance to have a non-carrier daughter, a 25% chance to have a diseased son, and a 25% son to have a healthy son.[10]
Signs and symptoms
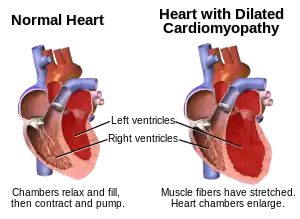
In general, the signs and symptoms of AVM are similar to common neuromuscular disorders,[12] including limb-girdle weakness, scapuloperoneal dystrophy, distal myopathy and cardiomyopathy.[13] Both muscles of upper and lower extremity would be affected.[3] The symptoms are progressive. Patients might start off having difficulty in buttoning their clothes and picking up tiny objects.[3] As it progresses to a more severe stage, they would have difficulty walking and rising up from the chair.[1] Peripheral neuropathy is exhibited in approximately 20% of patients, and cardiomyopathy affects 15-30% of AVM patients. The age of onset typically ranges from early childhood to late adulthood.[1]
In addition, patients of Danon disease also experience cardiomyopathy, arrhythmia, and skeletal myopathy.[1] Men may also have varying degrees of intellectual disability with additional symptoms being retinal, liver and pulmonary disease.[1] Carrier females may exhibit these symptoms as well.[1]
Patients with Pompe disease exhibit a similar set of symptoms, ranging from progressive debilitation, to organ failure and death, of which the severity depends on age of onset, organ involvement, and rate of progression.[14] The onset of Pompe disease varies from infantile, late-infantile, childhood, juvenile, and adult-onset, though Pompe disease is broadly classified into just infant and late-onset.[14] Specifically, for patients with the progressive form of infantile-onset Pompe disease (IOPD), their symptoms eventually result in their deaths due to cardiorespiratory failure as soon as their first year of developing the disease.[15]
XMEA exhibits a similar set of symptoms as other AVMs, though the age of onset is predominantly during childhood.[16]
Diagnosis
Diagnosis for genetic or rare diseases are often challenging. As such, diagnosis includes a combination of genetic testing, followed by a thorough examination of a person's medical history, symptoms, a physical exam, and laboratory tests.[4] In addition, a diagnosis may also include a biopsy of the affected muscle tissue.[1]
For example, the diagnosis of Pompe disease presents a serious diagnostic dilemma as a result of the rarity and nonspecific phenotypic features of Pompe disease.[15] In other words, the overlapping signs and symptoms that Pompe disease shares with other neuromuscular diseases, such as muscle weakness and even cardiovascular diseases, create significant challenges for diagnosis.[14] Muscle biopsy is often used as an early diagnostic tool in the diagnosis of all muscular diseases.[17] However, it has been discovered that the use of muscle biopsies alone tends to result in false-negative results and subsequent delays in identifying and treating Pompe disease.[17] Therefore, a combination of several tests is typically used in conjunction to confirm whether a patient has Pompe disease.[17]
As mentioned, the signs and symptoms of Pompe disease, such as poor muscle tone and an enlarged heart, are typical to other conditions too.[14] As such, a thorough and differential diagnosis by specialists can help to distinguish Pompe from other diseases with similar symptoms.[18] Supposing the differential diagnosis concludes that Pompe disease is indeed present, further testing such as enzyme activity tests to measure the levels and activity of the acid alpha-glucosidase enzyme, or genetic testing to examine the GAA gene to determine mutations, should be performed for confirmation.[19] In addition, chest x-rays to check for an enlarged heart, electrocardiograms (ECG) to detect abnormal heartbeat patterns, and electromyograms (EMG) to assess for the presence of muscle injury and dysfunction, are additional tests that are performed to diagnosis Pompe disease.[15]
Treatment
There are two major approaches to treating Autophagic Vacuolar Myopathy: Enzyme Replacement Therapy (ERT) to compensate for the deficiency of the original enzyme by adding exogenous enzyme;[20] and gene therapy to replace or add genes to the body so that the body can produce the required protein.[21] ERT has been the main form of treatment though its effect is temporary. While gene therapy could be the ultimate cure, it is still under active research and more time is required before it can be applied to patients of AVM.[7]
Enzyme replacement therapy (ERT)
Enzyme Replacement Therapy has been used to treat Pompe disease.[22] Since Pompe disease is caused by the deficiency of the enzyme acid alpha-glucosidase, the injection of the enzyme would alleviate the symptoms caused the disease.[20] The functional enzyme is produced by genetically modified cells in the laboratory.[20] It is then harvested, purified and eventually injected into the patient's body.[20] With this enzyme, the body will be able to convert glycogen to glucose, providing the body with energy and thus alleviating the problem of energy deficiency in the muscles and heart.[23] However, the exogenous enzyme might induce the production of neutralising antibodies, so immunosuppressive drugs are often consumed simultaneously to reduce the number of neutralising antibodies that could prevent the exogenous enzyme from exerting its effect.[24]
A major limitation of ERT is that injections are needed routinely, meaning that ERT may not be a permanent cure since it can only improve the patient's muscle strength in the short term.[25] Another constraint is that the enzyme is unable to cross the blood-brain barrier, meaning that ERT is unable to correct the neurological symptoms of the disease.[25]
Despite the clear clinical benefits, some side effects from ERT are exhibited. These included mild allergic reactions towards the enzyme, which may snowball into anaphylaxis, a life-threatening allergy.[26] Other side effects are headaches, nausea, vomiting, chest discomfort and abnormal blood pressure.[27]
Gene therapy
AVM are genetic diseases caused by mutations of the genes.[5] In this way, AVM can be addressed with gene therapy. Gene therapy starts with the repairing or replacement of the gene that causes the disease.[28] If this does not work, additional genes will be inserted into the body to address the mutated gene.[29] Danon Disease, the most common form of AVM, could possibly be cured by adeno-associated virus 9 (AAV9)–mediated gene therapy.[30] Clinical studies on mice with the mutated LAMP2 gene have shown that the injection of the normal human LAMP2B gene into Lamp2 knockout mice actually managed to cure the mice from Danon disease.[7] This method could be a permanent solution since the gene would be able to generate the protein needed and normal functions of the protein would be restored.[31] Nevertheless, this method has not yet been trialed on human subjects so it needs further clinical trials to show its validity. The same concept could be applied to other AVM, targeting different gene mutations by introducing the normal version of the mutated gene.[31]
References
- 1 2 3 4 5 6 7 8 9 "Congenital autophagic vacuolar myopathy is allelic to X-linked myopathy with excessive autophagy". Neurology. 93 (8): 371.2–371. August 2019. doi:10.1212/wnl.0000000000007478. PMC 7508314. PMID 31427494. S2CID 201093720.
- 1 2 "KEGG DISEASE: Autophagic vacuolar myopathy". www.genome.jp. Retrieved 2020-03-29.
- 1 2 3 Weihl CC, Iyadurai S, Baloh RH, Pittman SK, Schmidt RE, Lopate G, et al. (March 2015). "Autophagic vacuolar pathology in desminopathies". Neuromuscular Disorders. 25 (3): 199–206. doi:10.1016/j.nmd.2014.12.002. PMC 4355324. PMID 25557463.
- 1 2 "Test | Invitae Autophagic Vacuolar Myopathy Panel". www.invitae.com. Retrieved 2020-03-29.
- 1 2 3 4 5 6 7 8 9 Nishino I (January 2010). "[Eludication of pathomechanism of and development of therapy for autophagic vacuolar myopathies]". Rinsho Shinkeigaku = Clinical Neurology. 50 (1): 1–6. doi:10.5692/clinicalneurol.50.1. PMID 20120346.
- 1 2 Leah Plumb A (April 2004). "Genetics Home Reference". Reference Reviews. 18 (3): 38–39. doi:10.1108/09504120410528234. ISSN 0950-4125.
- 1 2 3 "Does Therapy Work and How?", Integrative Therapy: A Practitioner's Guide, SAGE Publications Ltd, pp. 23–44, 2007, doi:10.4135/9781446279892.n2, ISBN 978-1-4129-1211-2
- ↑ Johnson B (November 2007), "9. Voice, Power and Modernity", Talking and Listening in the Age of Modernity: Essays on the history of sound, ANU Press, doi:10.22459/tlam.11.2007.09, ISBN 978-1-921313-47-9
- ↑ "Sex Linked". Genome.gov. Retrieved 2020-04-22.
- 1 2 3 "Danon disease". Genetics Home Reference. Retrieved 2020-04-06.
- 1 2 3 4 5 6 "Danon Disease (Xq24)", Encyclopedic Dictionary of Genetics, Genomics and Proteomics, John Wiley & Sons, Inc., 2004-07-15, doi:10.1002/0471684228.egp03102, ISBN 0-471-68422-8
- ↑ Strehle EM (August 2009). "Food for thought: autophagic vacuolar myopathies". Archives of Disease in Childhood. 94 (8): 567–9. doi:10.1136/adc.2008.155010. PMID 19628877. S2CID 44794844.
- ↑ Millichap, J. Gordon (2011), "Limb Girdle Muscular Dystrophies", Pediatric Neurology Briefs, 17 (5): 39, doi:10.15844/pedneurbriefs-17-5-6
- 1 2 3 4 Bay LB, Denzler I, Durand C, Eiroa H, Frabasil J, Fainboim A, et al. (August 2019). "Infantile-onset Pompe disease: Diagnosis and management". Archivos Argentinos de Pediatria. 117 (4): 271–278. doi:10.5546/aap.2019.eng.271. PMID 31339275.
- 1 2 3 Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. (May 2006). "Pompe disease diagnosis and management guideline". Genetics in Medicine. 8 (5): 267–88. doi:10.1097/01.gim.0000218152.87434.f3. PMC 3110959. PMID 16702877.
- ↑ "X-Linked Myopathy with Excessive Autophagy". NORD (National Organization for Rare Disorders). Retrieved 2020-04-06.
- 1 2 3 Vissing J, Lukacs Z, Straub V (July 2013). "Diagnosis of Pompe disease: muscle biopsy vs blood-based assays". JAMA Neurology. 70 (7): 923–7. doi:10.1001/2013.jamaneurol.486. PMID 23649721.
- ↑ Dubrovsky A, Corderi J, Karasarides T, Taratuto AL (April 2013). "Pompe disease, the must-not-miss diagnosis: A report of 3 patients". Muscle & Nerve. 47 (4): 594–600. doi:10.1002/mus.23643. PMID 23463700. S2CID 5514255.
- ↑ Iskit S (3 May 2018). "Diagnosis of Pompe Disease". Pompe Disease News.
- 1 2 3 4 Bhui R, Spector AR (April 2020). "Obstructive sleep apnea in late-onset Pompe disease treated by enzyme replacement therapy". Neuromuscular Disorders. 30 (4): 329–330. doi:10.1016/j.nmd.2020.02.004. PMID 32173248. S2CID 211082029.
- ↑ Appleby CE, Kingston PA (June 2004). "Gene therapy for restenosis--what now, what next?". Current Gene Therapy. 4 (2): 153–82. doi:10.2174/1566523043346435. PMID 15180583.
- ↑ Johnson EM, Roberts M, Mozaffar T, Young P, Quartel A, Berger KI (February 2016). "Pulmonary function tests (maximum inspiratory pressure, maximum expiratory pressure, vital capacity, forced vital capacity) predict ventilator use in late-onset Pompe disease". Neuromuscular Disorders. 26 (2): 136–45. doi:10.1002/14651858.cd012993. PMC 6494567. PMID 26794303.
- ↑ Kuperus E, Kruijshaar ME, Wens SC, de Vries JM, Favejee MM, van der Meijden JC, et al. (December 2017). "Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study". Neurology. 89 (23): 2365–2373. doi:10.1212/WNL.0000000000004711. PMID 29117951. S2CID 22669525.
- ↑ Matzner U (2005). "Therapy of Lysosomal Storage Diseases". Lysosomes. Springer US. pp. 112–129. doi:10.1007/0-387-28957-7_10. ISBN 978-0-387-25562-0.
- 1 2 Schoenbach A (2018-09-10). "Enzyme Replacement Therapy". Pompe Disease News. Retrieved 2020-03-29.
- ↑ Ratner M (Aug 2009). "Genzyme's Lumizyme clears bioequivalence hurdles". Nature Biotechnology. 27 (8): 685. doi:10.1038/nbt0809-685a. ISSN 1087-0156. S2CID 9682328.
- ↑ "Lumizyme | About Lumizyme". home. Retrieved 2020-03-29.
- ↑ Hellman D (2017-09-08), "What Makes Genetic Discrimination Exceptional?", Genetics and Gene Therapy, Routledge, pp. 169–208, doi:10.4324/9781315254517-7, ISBN 978-1-315-25451-7
- ↑ "What is gene therapy?". Genetics Home Reference. Retrieved 2020-03-29.
- ↑ Manso AM, Hashem SI, Nelson BC, Gault E, Soto-Hermida A, Villarruel E, et al. (March 2020). "Systemic AAV9.LAMP2B injection reverses metabolic and physiologic multiorgan dysfunction in a murine model of Danon disease". Science Translational Medicine. 12 (535): eaax1744. doi:10.1126/scitranslmed.aax1744. PMID 32188720. S2CID 213193924.
- 1 2 "How does gene therapy work?". Genetics Home Reference. Retrieved 2020-03-29.