| BCKDHA | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | BCKDHA, BCKDE1A, MSU, MSUD1, OVD1A, branched chain keto acid dehydrogenase E1, alpha polypeptide, branched chain keto acid dehydrogenase E1 subunit alpha | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 608348 MGI: 107701 HomoloGene: 569 GeneCards: BCKDHA | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||



A 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial is an enzyme that in humans is encoded by the BCKDHA gene.[5]
BCKDHA is a coding gene that is part of the BCKD complex (branched-chain alpha-keto acid dehydrogenase).
Discovery
BCKDHA was discovered by John Menkes in 1954. After he had seen a family with four children die only a few months after birth, he found that their urine smelled sweet like maple syrup. While he was not the one to discover the specific gene, he did discover the maple syrup urine disease[6](MSUD). The BCKD complex is made up of three different catalytic pieces. It was in 1960 when Dancis discovered the gene itself, but this was from Menkes discovering of the disease leading to further investigation of its origin. He found that looking at the branched-chain amino acids and their corresponding alpha-keto acids in turn aided in the realization that they were pathogenetic compounds. Dancis was the one to specifically track down the enzymatic defect in (MSUD) by finding what gene in the pool of human chromosomes was defecting the urine. He found the gene on the level of the decarboxylation of the branched-chain amino acids.[7]
Gene location
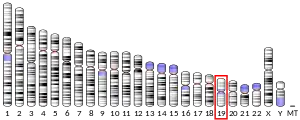
The cytogenetic location of BCKDHA is on the human chromosome 19, specifically on the cytogenetic band at 19q13.2. This the long arm (q) of the chromosome 19 at 13.2. Looking at the molecular location, the base pairs 41,397,789 to 41,425,005 are on chromosome 19. The cellular localization of this gene is within the mitochondrion matrix.[8]
Function
The second major step in the catabolism of the branched-chain amino acids (isoleucine, leucine, and valine) is catalyzed by the branched-chain alpha-keto acid dehydrogenase complex (BCKD; EC 1.2.4.4), an inner-mitochondrial enzyme complex that consists of 3 catalytic components: a heterotetrameric (alpha2, beta2) branched-chain alpha-keto acid decarboxylase (E1), a homo-24-meric dihydrolipoyl transacylase (E2; MIM 248610), and a homodimeric dihydrolipoamide dehydrogenase (E3; MIM 238331). The reaction is irreversible and constitutes the first committed step in BCAA oxidation. The complex also contains 2 regulatory enzymes, a kinase and a phosphorylase. The BCKDHA gene encodes the alpha subunit of E1, and the BCKDHB gene (MIM 248611) encodes the beta subunit of E1.[supplied by OMIM][5]
The normal function of the BCKDHA gene is to provide instructions for making the alpha subunit of the branched-chain alpha-keto dehydrogenase (BCKD) enzyme complex. The alpha subunit is one part of the BCKD enzyme complex. Two beta subunits are produced from the BCKDHB gene [9] and connect to two alpha subunits to form the E1 (decarboxylase) component. The BCKD enzyme complex catalyzes one step in breaking down amino acids. Those amino acids being leucine, isoleucine, and valine. The BCKD enzyme complex can be found in the mitochondria, an organelle known as the powerhouse of the cell. All three amino acids can be found in protein-rich foods and when broken down, they can be used for energy. Mutations in the BCKDHA gene can lead to maple syrup urine disease.[10]
Clinical significance
Mutations in the BCKDHA gene occur due to single point mutations in the “alpha subunit of the BCKD enzyme complex”.[6] Earlier cases of this disease show the mutation more frequently occurred by replacing the amino acid tyrosine. This amino acid was replaced with asparagine. The complication with mutations in the BCKDHA gene is that it disrupts the normal function of the BCKD enzyme complex, preventing the gene from going about its normal functions. Thus, the BCKDHA gene would not be able to break down leucine, isoleucine, and valine. When these byproducts start to accumulate it produces a toxic environment for cells and tissues, specifically in the nervous system. This can lead to seizures, developmental delay, but most importantly maple syrup urine disease.
The BCKDHA has been pinpointed in people with maple syrup urine disease, due to over 80 mutations occurring in that gene. Severe symptoms arise from these mutations and cause the disease which shows soon after birth. Due to the sweet odor from the urine, the disease was termed maple syrup urine disease. The disease causes loss of appetite, nausea, lethargy, and delayed development.
BCKDHA mutation: maple syrup urine disease
Maple syrup urine disease is an “autosomal recessive inborn error of metabolism. Meaning, as stated earlier, that there is a defect (i.e. error) in the single gene that codes for an enzyme. These enzymes promote conversions for various substrates into products. In terms of maple syrup urine disease, the enzyme defect occurs in the metabolic pathway of the “branched-chain amino acids” leucine, isoleucine, and valine.[9] The buildup of these amino acids lead to “encephalopathy and progressive neurodegeneration”;[9] along with other complications.
There are five forms of maple syrup urine disease: intermediate, intermittent, thiamine-responsive and E3 deficient. The form of disease is dependent upon clinical prognosis, dietary protein tolerance, and thiamine response and level of enzyme activity. Intermediate maple syrup urine disease is a milder form of maple syrup urine disease because it persistently raises branched-chain amino acids and some keto-acid chains. Individuals with this disease have a partial BCKDHA enzyme deficiency.[6] Meaning that it shows up sporadically or reacts to dietary thiamine therapy.
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000248098 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000060376 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- 1 2 "Entrez Gene: BCKDHA branched chain keto acid dehydrogenase E1, alpha polypeptide".
- 1 2 3 Reference, Genetics Home. "BCKDHA gene". Genetics Home Reference. Retrieved 2018-11-14.
- ↑ Reference, Genetics Home. "BCKDHA gene". Genetics Home Reference. Retrieved 2018-11-15.
- ↑ "BCKDHA branched chain keto acid dehydrogenase E1, alpha polypeptide". NCBI. Retrieved 12 April 2019.
- 1 2 3 Database, GeneCards Human Gene. "BCKDHA Gene - GeneCards | ODBA Protein | ODBA Antibody". www.genecards.org. Retrieved 2018-11-14.
- ↑ Database, GeneCards Human Gene. "BCKDHA Gene - GeneCards | ODBA Protein | ODBA Antibody". www.genecards.org. Retrieved 2018-11-15.
Further reading
- Mitsubuchi H, Matsuda I, Nobukuni Y, Heidenreich R, Indo Y, Endo F, Mallee J, Segal S (1992). "Gene analysis of Mennonite maple syrup urine disease kindred using primer-specified restriction map modification". Journal of Inherited Metabolic Disease. 15 (2): 181–7. doi:10.1007/BF01799628. PMID 1356170. S2CID 35446485.
- McKean MC, Winkeler KA, Danner DJ (November 1992). "Nucleotide sequence of the 5' end including the initiation codon of cDNA for the E1 alpha subunit of the human branched chain alpha-ketoacid dehydrogenase complex". Biochimica et Biophysica Acta. 1171 (1): 109–12. doi:10.1016/0167-4781(92)90149-t. PMID 1420356.
- Dariush N, Fisher CW, Cox RP, Chuang DT (October 1991). "Structure of the gene encoding the entire mature E1 alpha subunit of human branched-chain alpha-keto acid dehydrogenase complex (1991) FEBS Letters 284, 34-38". FEBS Letters. 291 (2): 376–7. doi:10.1016/0014-5793(91)81324-2. PMID 1682165.
- Eisenstein RS, Hoganson G, Miller RH, Harper AE (1991). "Altered phosphorylation state of branched-chain 2-oxo acid dehydrogenase in a branched-chain acyltransferase deficient human fibroblast cell line". Journal of Inherited Metabolic Disease. 14 (1): 37–44. doi:10.1007/BF01804386. PMID 1861457. S2CID 25819969.
- Fisher CR, Fisher CW, Chuang DT, Cox RP (August 1991). "Occurrence of a Tyr393----Asn (Y393N) mutation in the E1 alpha gene of the branched-chain alpha-keto acid dehydrogenase complex in maple syrup urine disease patients from a Mennonite population". American Journal of Human Genetics. 49 (2): 429–34. PMC 1683290. PMID 1867199.
- Fisher CR, Chuang JL, Cox RP, Fisher CW, Star RA, Chuang DT (September 1991). "Maple syrup urine disease in Mennonites. Evidence that the Y393N mutation in E1 alpha impedes assembly of the E1 component of branched-chain alpha-keto acid dehydrogenase complex". The Journal of Clinical Investigation. 88 (3): 1034–7. doi:10.1172/JCI115363. PMC 295513. PMID 1885764.
- Zneimer SM, Lau KS, Eddy RL, Shows TB, Chuang JL, Chuang DT, Cox RP (July 1991). "Regional assignment of two genes of the human branched-chain alpha-keto acid dehydrogenase complex: the E1 beta gene (BCKDHB) to chromosome 6p21-22 and the E2 gene (DBT) to chromosome 1p31". Genomics. 10 (3): 740–7. doi:10.1016/0888-7543(91)90458-Q. PMID 1889817.
- Zhang B, Zhao Y, Harris RA, Crabb DW (February 1991). "Molecular defects in the E1 alpha subunit of the branched-chain alpha-ketoacid dehydrogenase complex that cause maple syrup urine disease". Molecular Biology & Medicine. 8 (1): 39–47. PMID 1943689.
- Dariush N, Fisher CW, Cox RP, Chuang DT (June 1991). "Structure of the gene encoding the entire mature E1 alpha subunit of human branched-chain alpha-keto acid dehydrogenase complex". FEBS Letters. 284 (1): 34–8. doi:10.1016/0014-5793(91)80755-R. PMID 2060625. S2CID 83480996.
- Matsuda I, Nobukuni Y, Mitsubuchi H, Indo Y, Endo F, Asaka J, Harada A (October 1990). "A T-to-A substitution in the E1 alpha subunit gene of the branched-chain alpha-ketoacid dehydrogenase complex in two cell lines derived from Menonite maple syrup urine disease patients". Biochemical and Biophysical Research Communications. 172 (2): 646–51. doi:10.1016/0006-291X(90)90723-Z. PMID 2241958.
- Zhang B, Edenberg HJ, Crabb DW, Harris RA (April 1989). "Evidence for both a regulatory mutation and a structural mutation in a family with maple syrup urine disease". The Journal of Clinical Investigation. 83 (4): 1425–9. doi:10.1172/JCI114033. PMC 303839. PMID 2703538.
- Fekete G, Plattner R, Crabb DW, Zhang B, Harris RA, Heerema N, Palmer CG (1989). "Localization of the human gene for the El alpha subunit of branched chain keto acid dehydrogenase (BCKDHA) to chromosome 19q13.1----q13.2". Cytogenetics and Cell Genetics. 50 (4): 236–7. doi:10.1159/000132768. PMID 2805821.
- Fisher CW, Chuang JL, Griffin TA, Lau KS, Cox RP, Chuang DT (February 1989). "Molecular phenotypes in cultured maple syrup urine disease cells. Complete E1 alpha cDNA sequence and mRNA and subunit contents of the human branched chain alpha-keto acid dehydrogenase complex". The Journal of Biological Chemistry. 264 (6): 3448–53. doi:10.1016/S0021-9258(18)94087-1. PMID 2914958.
- Zhang B, Crabb DW, Harris RA (September 1988). "Nucleotide and deduced amino acid sequence of the E1 alpha subunit of human liver branched-chain alpha-ketoacid dehydrogenase". Gene. 69 (1): 159–64. doi:10.1016/0378-1119(88)90390-3. PMID 3224821.
- Hu CW, Lau KS, Griffin TA, Chuang JL, Fisher CW, Cox RP, Chuang DT (June 1988). "Isolation and sequencing of a cDNA encoding the decarboxylase (E1)alpha precursor of bovine branched-chain alpha-keto acid dehydrogenase complex. Expression of E1 alpha mRNA and subunit in maple-syrup-urine-disease and 3T3-L1 cells". The Journal of Biological Chemistry. 263 (18): 9007–14. doi:10.1016/S0021-9258(18)68408-X. PMID 3379058.
- Chuang JL, Davie JR, Chinsky JM, Wynn RM, Cox RP, Chuang DT (March 1995). "Molecular and biochemical basis of intermediate maple syrup urine disease. Occurrence of homozygous G245R and F364C mutations at the E1 alpha locus of Hispanic-Mexican patients". The Journal of Clinical Investigation. 95 (3): 954–63. doi:10.1172/JCI117804. PMC 441427. PMID 7883996.
- Wynn RM, Kochi H, Cox RP, Chuang DT (September 1994). "Differential processing of human and rat E1 alpha precursors of the branched-chain alpha-keto acid dehydrogenase complex caused by an N-terminal proline in the rat sequence". Biochimica et Biophysica Acta (BBA) - General Subjects. 1201 (1): 125–8. doi:10.1016/0304-4165(94)90161-9. PMID 7918575.
- Chuang JL, Fisher CR, Cox RP, Chuang DT (August 1994). "Molecular basis of maple syrup urine disease: novel mutations at the E1 alpha locus that impair E1(alpha 2 beta 2) assembly or decrease steady-state E1 alpha mRNA levels of branched-chain alpha-keto acid dehydrogenase complex". American Journal of Human Genetics. 55 (2): 297–304. PMC 1918348. PMID 8037208.
- Nobukuni Y, Mitsubuchi H, Hayashida Y, Ohta K, Indo Y, Ichiba Y, Endo F, Matsuda I (November 1993). "Heterogeneity of mutations in maple syrup urine disease (MSUD): screening and identification of affected E1 alpha and E1 beta subunits of the branched-chain alpha-keto-acid dehydrogenase multienzyme complex". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1225 (1): 64–70. doi:10.1016/0925-4439(93)90123-i. PMID 8161368.
- Bonaldo MF, Lennon G, Soares MB (September 1996). "Normalization and subtraction: two approaches to facilitate gene discovery". Genome Research. 6 (9): 791–806. doi:10.1101/gr.6.9.791. PMID 8889548.
External links
- Human BCKDHA genome location and BCKDHA gene details page in the UCSC Genome Browser.
PDB gallery | |
|---|---|
|