Brunauer–Emmett–Teller (BET) theory aims to explain the physical adsorption of gas molecules on a solid surface and serves as the basis for an important analysis technique for the measurement of the specific surface area of materials. The observations are very often referred to as physical adsorption or physisorption. In 1938, Stephen Brunauer, Paul Hugh Emmett, and Edward Teller presented their theory in the Journal of the American Chemical Society.[1] BET theory applies to systems of multilayer adsorption that usually utilizes a probing gas (called the adsorbate) that does not react chemically with the adsorptive (the material upon which the gas attaches to) to quantify specific surface area. Nitrogen is the most commonly employed gaseous adsorbate for probing surface(s). For this reason, standard BET analysis is most often conducted at the boiling temperature of N2 (77 K). Other probing adsorbates are also utilized, albeit less often, allowing the measurement of surface area at different temperatures and measurement scales. These include argon, carbon dioxide, and water. Specific surface area is a scale-dependent property, with no single true value of specific surface area definable, and thus quantities of specific surface area determined through BET theory may depend on the adsorbate molecule utilized and its adsorption cross section.[2]
Concept

The concept of the theory is an extension of the Langmuir theory, which is a theory for monolayer molecular adsorption, to multilayer adsorption with the following hypotheses:
- gas molecules physically adsorb on a solid in layers infinitely;
- gas molecules only interact with adjacent layers; and
- the Langmuir theory can be applied to each layer.
- the enthalpy of adsorption for the first layer is constant and greater than the second (and higher).
- the enthalpy of adsorption for the second (and higher) layers is the same as the enthalpy of liquefaction.
The resulting BET equation is

where c is referred to as the BET C-constant, is the vapor pressure of the adsorptive bulk liquid phase which would be at the temperature of the adsorbate and θ is the surface coverage, defined as:

.

Here is the amount of adsorbate and is called the monolayer equivalent. The is the entire amount that would be present as a monolayer (which is theoretically impossible for physical adsorption) would cover the surface with exactly one layer of adsorbate. The above equation is usually rearranged to yield the following equation for the ease of analysis:


![{\displaystyle {\frac {{p}/{p_{0}}}{v\left[1-\left({p}/{p_{0}}\right)\right]}}={\frac {c-1}{v_{\mathrm {m} }c}}\left({\frac {p}{p_{0}}}\right)+{\frac {1}{v_{m}c}},\qquad (1)}](../I/0b4d4a74db2ffb1f5e5a069deb6556899f951bbe.svg)
where and are the equilibrium and the saturation pressure of adsorbates at the temperature of adsorption, respectively; is the adsorbed gas quantity (for example, in volume units) while is the monolayer adsorbed gas quantity. is the BET constant,






where is the heat of adsorption for the first layer, and is that for the second and higher layers and is equal to the heat of liquefaction or heat of vaporization.


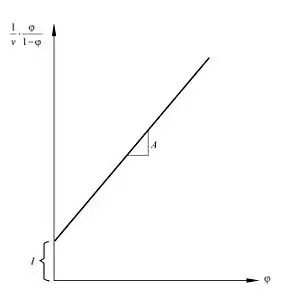
Equation (1) is an adsorption isotherm and can be plotted as a straight line with on the y-axis and on the x-axis according to experimental results. This plot is called a BET plot. The linear relationship of this equation is maintained only in the range of . The value of the slope and the y-intercept of the line are used to calculate the monolayer adsorbed gas quantity and the BET constant . The following equations can be used:
![{\displaystyle 1/{v[({p_{0}}/{p})-1]}}](../I/d345b7eea3cf3b66aa99e6972e208fba4fb3e3c0.svg)






The BET method is widely used in materials science for the calculation of surface areas of solids by physical adsorption of gas molecules. The total surface area and the specific surface area are given by




where is in units of volume which are also the units of the monolayer volume of the adsorbate gas, is the Avogadro number, the adsorption cross section of the adsorbate,[3] the molar volume of the adsorbate gas, and the mass of the solid sample or adsorbent.




Derivation
The BET theory can be derived similarly to the Langmuir theory, but by considering multilayered gas molecule adsorption, where it is not required for a layer to be completed before an upper layer formation starts. Furthermore, the authors made five assumptions:[4]
- Adsorptions occur only on well-defined sites of the sample surface (one per molecule)
- The only molecular interaction considered is the following one: a molecule can act as a single adsorption site for a molecule of the upper layer.
- The uppermost molecule layer is in equilibrium with the gas phase, i.e. similar molecule adsorption and desorption rates.
- The desorption is a kinetically limited process, i.e. a heat of adsorption must be provided:
- these phenomena are homogeneous, i.e. same heat of adsorption for a given molecule layer.
- it is E1 for the first layer, i.e. the heat of adsorption at the solid sample surface
- the other layers are assumed similar and can be represented as condensed species, i.e. liquid state. Hence, the heat of adsorption is EL is equal to the heat of liquefaction.
- At the saturation pressure, the molecule layer number tends to infinity (i.e. equivalent to the sample being surrounded by a liquid phase)
Consider a given amount of solid sample in a controlled atmosphere. Let θi be the fractional coverage of the sample surface covered by a number i of successive molecule layers. Let us assume that the adsorption rate Rads,i-1 for molecules on a layer (i-1) (i.e. formation of a layer i) is proportional to both its fractional surface θi-1 and to the pressure P, and that the desorption rate Rdes,i on a layer i is also proportional to its fractional surface θi:


where ki and k−i are the kinetic constants (depending on the temperature) for the adsorption on the layer (i−1) and desorption on layer i, respectively. For the adsorptions, these constants are assumed similar whatever the surface. Assuming an Arrhenius law for desorption, the related constants can be expressed as

where Ei is the heat of adsorption, equal to E1 at the sample surface and to EL otherwise.
Consider some substance A. The adsorption of A onto an available surface site produces a new site on the first layer. In summary,




Extending this to higher order layers one obtains

and similarly



Denoting the activity of the number of available sites of the th layer with and the partial pressure of A with , the last equilibrium can be written




It follows that the coverage of the first layer can be written

and that the coverage of the second layer can be written

Realising that the adsorption of A onto the second layer is equivalent to adsorption of A onto its own liquid phase,[5] the rate constant for should be the same, which results in the recursion


In order to simplify some infinite summations, let and let . Then the th layer coverage can written



if . The coverage of any layer is defined as the relative number of available sites. An alternative definition, which leads to a set of coverage's that are numerically to those resulting from the original way of defining surface coverage, is that denotes the relative number of sites covered by only adsorbents.[5] Doing so it is easy to see that the total volume of adsorbed molecules can be written as the sum


where is the molecular volume. Employing the fact that this sum is the first derivative of a geometric sum, the volume becomes


Since the total coverage of a mono-layer must be unity, the full mono-layer coverage must be

In order to properly make the substitution for , the restriction forces us to take the zeroth contribution outside the summation, resulting in

Lastly, defining the excess coverage as , the excess volume relative to the volume of an adsorbed mono-layer becomes


where the last equality was obtained by making use of the series expansions presented above. The constant must be interpreted as the relative binding affinity the substance A has towards a surface, relative to its own liquid. If then the initial part of the isotherm will be reminiscent of the Langmuir isotherm which reaches a plateau at full mono-layer coverage, whereas means the mono-layer will have a slow build-up. Another thing to note is that in order for the geometric substitutions to hold, . The isotherm above exhibits a singularity at . Since one can write , implying that . This means that must be true, ultimately resulting in .








Finding the linear BET range
It is still not clear on how to find the linear range of the BET plot for microporous materials in a way that reduces any subjectivity in the assessment of the monolayer capacity. A crowd-sourced study involving 61 research groups has shown that reproducibility of BET area determination from identical isotherms is, in some cases, problematic.[6] Rouquerol et al.[7] suggested a procedure that is based on two criteria:
- C must be positive implying that any negative intercept on the BET plot indicates that one is outside the valid range of the BET equation.
- Application of the BET equation must be limited to the range where the term V(1-P/P0) continuously increases with P/P0.
These corrections are an attempt to salvage the BET theory, which is restricted to type II isotherms. Even while using this type, use of the data itself is restricted to 0.05 to 0.35 of , routinely discarding 70% of the data. This restriction must be modified depending upon conditions.

Limitations of BET
Terrell L. Hill described BET as a theory that is "... extremely useful as a qualitative guide; but it is not quantitatively correct".[8] Although BET adsorption isotherm is still extensively used for different applications and is used for specific surface area determinations of powders whose calculation is not sensitive to the simplifications of the BET theory.[8] Both Hackerman's[9] and Sing's group[10] have highlighted the limitations of the BET method.[11] Hackerman et al. noted the potential for 10% uncertainty in the method's values,[9] with Sing's group attributed the significant variation in reported values of molecular area to the BET method's possible inaccurate assessment of monolayer capacity.[11] In subsequent studies using the BET interpretation of nitrogen and water vapor adsorption isotherms, the reported area occupied by an adsorbed water molecule on fully hydroxylated silica ranged from 0.25 to 0.44 nm².[11] Other issues with the BET include the fact that in certain cases, BET leads to anomalies such as reaching an infinite amount adsorbed at reaching unity,[12] and in some cases, the constant C (surface binding energy) can be determined to be negative.[13]
Applications
Cement and concrete
The rate of curing of concrete depends on the fineness of the cement and of the components used in its manufacture, which may include fly ash, silica fume and other materials, in addition to the calcinated limestone which causes it to harden. Although the Blaine air permeability method is often preferred, due to its simplicity and low cost, the nitrogen BET method is also used.
When hydrated cement hardens, the calcium silicate hydrate (or C-S-H), which is responsible for the hardening reaction, has a large specific surface area because of its high porosity. This porosity is related to a number of important properties of the material, including the strength and permeability, which in turn affect the properties of the resulting concrete. Measurement of the specific surface area using the BET method is useful for comparing different cements. This may be performed using adsorption isotherms measured in different ways, including the adsorption of water vapour at temperatures near ambient, and adsorption of nitrogen at 77 K (the boiling point of liquid nitrogen). Different methods of measuring cement paste surface areas often give very different values, but for a single method the results are still useful for comparing different cements.
Activated carbon
Activated carbon has strong affinity for many gases and has an adsorption cross section of 0.162 nm2 for nitrogen adsorption at liquid-nitrogen temperature (77 K). BET theory can be applied to estimate the specific surface area of activated carbon from experimental data, demonstrating a large specific surface area, even around 3000 m2/g.[14] However, this surface area is largely overestimated due to enhanced adsorption in micropores,[7] and more realistic methods should be used for its estimation, such as the subtracting pore effect (SPE) method.[15]
Catalysis
In the field of solid catalysis, the surface area of catalysts is an important factor in catalytic activity. Inorganic materials such as mesoporous silica and layered clay minerals have high surface areas of several hundred m2/g calculated by the BET method, indicating the possibility of application for efficient catalytic materials.
Specific surface area calculation
The ISO 9277 standard for calculating the specific surface area of solids is based on the BET method.
Thermal desorption
In 2023, researchers in the United States developed a method to determine BET surface areas using a thermogravimetric analyzer (TGA).[16] This method uses a TGA to heat a porous sample loaded with an adsorbate, the produced plot of sample mass vs. temperature is then mapped into a standard isotherm to which BET theory is applied as normal. Common fluids, e.g. water or toluene, can be used as adsorbates for the TGA method allowing the specific interactions of different adsorbates to be determined, as these frequently differ from the commonly used nitrogen.
See also
References
- ↑ Brunauer, Stephen; Emmett, P. H.; Teller, Edward (1938). "Adsorption of Gases in Multimolecular Layers". Journal of the American Chemical Society. 60 (2): 309–319. Bibcode:1938JAChS..60..309B. doi:10.1021/ja01269a023. ISSN 0002-7863.
- ↑ Hanaor, D. A. H.; Ghadiri, M.; Chrzanowski, W.; Gan, Y. (2014). "Scalable Surface Area Characterization by Electrokinetic Analysis of Complex Anion Adsorption" (PDF). Langmuir. 30 (50): 15143–15152. arXiv:2106.03411. doi:10.1021/la503581e. PMID 25495551. S2CID 4697498.
- ↑ Galarneau, Anne; Mehlhorn, Dirk; Guenneau, Flavien; Coasne, Benoit; Villemot, Francois; Minoux, Delphine; Aquino, Cindy; Dath, Jean-Pierre (2018-10-31). "Specific Surface Area Determination for Microporous/Mesoporous Materials: The Case of Mesoporous FAU-Y Zeolites" (PDF). Langmuir. American Chemical Society (ACS). 34 (47): 14134–14142. doi:10.1021/acs.langmuir.8b02144. ISSN 0743-7463. PMID 30379547. S2CID 53197261.
- ↑ Sing, Kenneth S.W. (1998). "Adsorption methods for the characterization of porous materials". Advances in Colloid and Interface Science. 76–77: 3–11. doi:10.1016/S0001-8686(98)00038-4.
- 1 2 Brunauer, Stephen; Emmett, P. H.; Teller, Edward (February 1938). "Adsorption of Gases in Multimolecular Layers". Journal of the American Chemical Society. 60 (2): 309–319. doi:10.1021/ja01269a023.
- ↑ Osterrieth, Johannes W. M.; Rampersad, James; Madden, David; Rampal, Nakul; Skoric, Luka; Connolly, Bethany; Allendorf, Mark D.; Stavila, Vitalie; Snider, Jonathan L.; Ameloot, Rob; Marreiros, João (2022-05-23). "How Reproducible are Surface Areas Calculated from the BET Equation?". Advanced Materials. 34 (27): 2201502. Bibcode:2022AdM....3401502O. doi:10.1002/adma.202201502. hdl:10754/678181. ISSN 0935-9648. PMID 35603497. S2CID 236753643.
- 1 2 Rouquerol, J.; Llewellyn, P.; Rouquerol, F. (2007), "Is the bet equation applicable to microporous adsorbents?", Studies in Surface Science and Catalysis, Elsevier, vol. 160, pp. 49–56, doi:10.1016/s0167-2991(07)80008-5, ISBN 9780444520227
- 1 2 Hill, Terrell L. (1952-01-01), Frankenburg, W. G.; Komarewsky, V. I.; Rideal, E. K. (eds.), "Theory of Physical Adsorption", Advances in Catalysis, Academic Press, vol. 4, pp. 211–258, doi:10.1016/s0360-0564(08)60615-x, retrieved 2023-07-22
- 1 2 Makrides, A. C.; Hackerman, Norman (April 1959). "Heats of Immersion. I. The System Silica–water". The Journal of Physical Chemistry. 63 (4): 594–598. doi:10.1021/j150574a035. ISSN 0022-3654.
- ↑ Baker, Frederick S; Sing, Kenneth S.W (June 1976). "Specificity in the adsorption of nitrogen and water on hydroxylated and dehydroxylated silicas". Journal of Colloid and Interface Science. 55 (3): 605–613. doi:10.1016/0021-9797(76)90071-0. ISSN 0021-9797.
- 1 2 3 Narayanaswamy, Nagarajan; Ward, C. A. (2020-03-27). "Area Occupied by a Water Molecule Adsorbed on Silica at 298 K: Zeta Adsorption Isotherm Approach". The Journal of Physical Chemistry C. 124 (17): 9269–9280. doi:10.1021/acs.jpcc.9b11976. ISSN 1932-7447.
- ↑ Saber, Sepehr; Narayanaswamy, Nagarajan; Ward, C. A.; Elliott, Janet A. W. (2023-05-28). "Experimental examination of the phase transition of water on silica at 298 K". The Journal of Chemical Physics. 158 (20). doi:10.1063/5.0145932. ISSN 0021-9606.
- ↑ Al-Ghouti, Mohammad A.; Da'ana, Dana A. (July 2020). "Guidelines for the use and interpretation of adsorption isotherm models: A review". Journal of Hazardous Materials. 393: 122383. doi:10.1016/j.jhazmat.2020.122383. ISSN 0304-3894.
- ↑ Nakayama, Atsuko; Suzuki, Kazuya; Enoki, Toshiaki; Koga, Kei-ichi; Endo, Morinobu; Shindo, Norifumi (1996). "Electronic and Magnetic Properties of Activated Carbon Fibers". Bull. Chem. Soc. Jpn. 69 (2): 333–339. doi:10.1246/bcsj.69.333. ISSN 0009-2673. Retrieved 2015-06-26.
- ↑ Kaneko, K.; Ishii, C.; Ruike, M.; Kuwabara, H. (1992). "Origin of superhigh surface area and microcrystalline graphitic structures of activated carbons". Carbon. 30 (7): 1075–1088. doi:10.1016/0008-6223(92)90139-N. ISSN 0008-6223.
- ↑ Stahlfeld, K.; Belmont, E. (2023). "BET and Kelvin Analyses by Thermogravimetric Desorption". Langmuir. 39 (25): 8814–8823. doi:10.1021/acs.langmuir.3c00854. ISSN 0743-7463.