| Hereditary angioedema (HAE) | |
|---|---|
| Other names | Hereditary angioneurotic edema (HANE),[1] familial angioneurotic edema[2] |
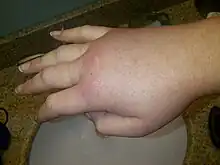 | |
| Swollen right hand during a hereditary angioedema attack. | |
| Specialty | Hematology |
| Symptoms | Recurrent attacks of severe swelling[3] |
| Usual onset | Childhood[3] |
| Duration | Attacks last a few days[3] |
| Types | Type I, II, III[3] |
| Causes | Genetic disorder (autosomal dominant)[3] |
| Diagnostic method | Measuring C4 and C1-inhibitor levels.[2] |
| Differential diagnosis | Intestinal obstruction, other types of angioedema[2] |
| Prevention | C1 inhibitor[1] |
| Treatment | Supportive care, medications[1] |
| Medication | C1 inhibitor, ecallantide, icatibant[1] |
| Prognosis | 25% risk of death if airway involved (without treatment)[2] |
| Frequency | ~1 in 50,000[3] |
Hereditary angioedema (HAE) is a disorder that results in recurrent attacks of severe swelling.[3] The swelling most commonly affects the arms, legs, face, intestinal tract, and airway.[3] If the intestinal tract is affected, abdominal pain and vomiting may occur.[1] Swelling of the airway can result in its obstruction and trouble breathing.[1] Without preventive treatment, attacks typically occur every two weeks and last for a few days.[3]
There are three main types of HAE.[3] Types I and II are caused by a mutation in the SERPING1 gene, which encodes the C1 inhibitor protein, while type III is often due to a mutation in the F12 (factor XII) gene.[3] The result is increased levels of bradykinin, which promotes swelling.[3] The condition may be inherited in an autosomal dominant manner or occur as a new mutation.[3] Triggers for an attack may include minor trauma or stress, but attacks often occur without any obvious preceding event.[3] Diagnosis of types I and II is based on measurement of C4 and C1-inhibitor levels.[2]
Management of HAE involves efforts to prevent attacks and the treatment of attacks if they occur.[1] During an attack, supportive care such as intravenous fluids and airway support may be required.[1] C1 inhibitor medications can be used for both prevention and treatment, while ecallantide and icatibant can be used to treat acute attacks.[1]
HAE affects approximately 1 in 50,000 people.[3] The condition is typically first noticed in childhood.[3] Type I and II affected females and males equally,[4] while type III affects females more often than males.[2] When the airway is involved, without treatment, the risk of death is about 25%.[2] With treatment, outcomes are generally good.[2] The condition was first described in 1888 by Canadian physician William Osler.[5]
Signs and symptoms
People diagnosed with Hereditary Angioedema have recurrent swelling in the extremities, genitals, face, lips, larynx or GI tract.[6] Some patients describe a sensation of fullness but not pain or itching in the affected area except for those with abdominal swellings who often experience acute abdominal pain. Others experience an intense amount of pain, described as radiating from the bone outward along with intense itching just beneath the skin and intense heat, regardless of the area targeted.
Swelling involving the respiratory and gastrointestinal systems can cause significant risk and distress. Involvement of respiratory structures, such as the throat or larynx, can cause difficulties in breathing and life-threatening airway obstruction.[6][7] Episodes that attack the gastrointestinal tract can cause a number of complications including vomiting, crampy abdominal pain, diarrhea, and dehydration.[7]
Some people with HAE experience 'wandering' attacks. These attacks will center around an extremity. For example: Should the affected person's hand swell up, it will go through the normal swelling cycle before 'transferring' to either the connection limb (In this case wrist to forearm) or move to the opposite hand. People with this symptom may find their episodes last longer and may find their triggers more difficult to track.
Some people may experience prodromal symptoms, including tingling, fatigue or weakness at the site of impending edema with one third of people experiencing an erythematous rash (erythema marginatum) as the prodromal symptom.[6] Urticaria is usually not seen in hereditary angioedema, as compared to other causes of angioedema such as histamine induced symptoms.[6]
Genetics
Because HAE is an autosomal dominant disease, there is no sex difference in transmission and both sexes are equally likely to receive the mutated gene from their parents. The autosomal dominant inheritance pattern with regards to hereditary angioedema requires receipt of only one copy of the mutated C1 inhibitor gene to have symptomatic disease.[8][9][10][11] Further, hereditary angioedema with C1 inhibitor deficiency types 1 and 2 have complete penetrance, meaning all of those who inherit the dysfunctional gene will have symptomatic disease. However, hereditary angioedema with normal C1 inhibitor levels (Type 3 disease) has incomplete penetrance, and men may be asymptomatic carriers despite inheriting a mutated gene.[6]
With regards to the mutations in the SERPING1 gene that is seen in hereditary angioedema types 1 and 2 (hereditary angioedema with C1 inhibitor deficiency), 75% of the cases are due to an autosomal dominant inheritance of a mutated gene and 25% of cases are due to de novo mutations of the egg or sperm, or early in embryological development.[6] Hereditary angioedema type 3 (hereditary angioedema with normal C1 inhibitor levels) is associated with mutations in genes for Factor XII, angiopoietin 1, plasminogen or kininogen 1.[6]
Pathophysiology
The pathophysiologic mechanisms contributing to bradykinin mediated angioedema in hereditary angioedema have been described. C1 inhibitor usually acts as an inhibitor of the plasma contact system. However in hereditary angioedema with C1 inhibitor deficiency, C1 inhibitor is either reduced in quantity and function (type 1) or dysfunctional (type 2), this leads to bradykinin disinhibition and bradykinin mediated activation of bradykinin B1 receptor and bradykinin B2 receptor on endothelial cells (cells lining blood vessels).[6] This activation leads to vascular endothelial cadherin (a type of cell adhesion molecule) phosphorylation, internalization and degradation. Cadherin degradation leads to actin cytoskeleton contraction and increased pore size of the vascular endothelial cells. Adherens junctions are also reduced due to bradykinin B1 and B2 receptor activation and vascular endothelial growth factor (VEGF) is also activated. The degradation of these endothelial intercellular barrier junctions mediated by histamine leads to increased vascular permeability, leading to vascular leakage into surrounding tissues and thus causing the characteristic swelling seen in hereditary angioedema.[6] The bradykinin B1 receptor (unlike the B2 receptor) is slowly and only partially desensitized after binding the bradykinin agonist, thus remaining consitutively active long after initial bradykinin exposure which can explain the protracted swelling seen in hereditary angioedema as compared to other causes of angioedema.[6]
Diagnosis
| C4 (C) | FB (A) | C3 | CH50 | Conditions |
|---|---|---|---|---|
| · | ↓ | ↓ | ↓ | PSG, C3 NeF AA |
| ↓ | · | ↓ | · | HAE, C4D |
| · | · | · | ↓ | TCPD |
| ↓ | ·/↓ | ↓ | ↓ | SLE |
| ↑ | ↑ | ↑ | ↑ | inflammation |
Recognizing HAE is often difficult due to the wide variability in disease expression, with the diagnosis often delayed for years.[6] The course of the disease is diverse and unpredictable, even within a single patient over their lifetime. This disease may be similar in its presentation to other forms of angioedema resulting from allergies or other medical conditions, but it is significantly different in cause and treatment. When HAE is misdiagnosed as an allergy it is most commonly treated with steroids, epinephrine or anti-histamines, drugs that are usually ineffective in treating a HAE episode.[6] Other misdiagnoses have resulted in unnecessary exploratory surgery for patients with abdominal swelling and other HAE patients report that their abdominal pain was wrongly diagnosed as psychosomatic or malingering.[6]
HAE accounts for only a small fraction of all cases of angioedema. To avoid potentially fatal consequences such as upper airway obstruction and unnecessary abdominal surgery, the importance of a correct diagnosis cannot be overemphasized.[12]
HAE should be considered if a patient presents with:
- Recurrent angioedema (without urticaria)
- Recurrent episodes of abdominal pain and vomiting
- Laryngeal edema
- Positive family history of angioedema[13][14]
A blood test, ideally taken during an episode, can be used to diagnose the condition. Measure: serum complement factor 4 (C4), C1 inhibitor (C1-INH) antigenic protein, C1 inhibitor (C1-INH) functional level if available. Analysis of complement C1 inhibitor levels may play a role in diagnosis. C4 and C2 are complementary components.
Types
There are three types of hereditary angioedema (HAE). HAE types I and II are both caused by a deficiency of complement C1-inhibitor (C1-INH), a plasma protein that is an important inhibitor of several serine proteases, specially of the complement system and the contact activation/kallikrein-kinin pathway, but also the fibrinolytic system.[15][16][17]
In HAE type I, there is a quantitative C1-inhibitor deficiency, antigenic as well as functional C1-inhibitor levels in plasma are decreased. This type accounts for approximately 85% of HAE cases with C1-inhibitor deficiency.[6]
In HAE type II, there is a qualitative deficiency, with normal - sometimes even elevated - C1-inhibitor protein levels, but decreased functional C1-inhibitor measurements. This type is seen in approximately 15% of HAE cases with C1-inhibitor deficiency.[17][18]
C1-inhibitor deficiency is caused by mutations of the SERPING1 gene, the gene encoding complement C1-inhibitor. More than 700 different mutations have been described.[18] The SERPING1 gene shows a considerable tendency for de novo mutations. As a result, it is not uncommon to observe patients with primary recurrent angioedema attacks and C1-inhibitor deficiency where both parents are unaffected.[19]
Another type of hereditary angioedema, originally named HAE type III, has been observed.[20][21][22] In contrast to HAE types I and II, this type of the disease is characterized by normal C1-inhibitor measurements. Thus, the term "hereditary angioedema with normal C1-inhibitor" is now generally used for this HAE type. Normal C1 inhibitor level hereditary angioedema is thought to involve various mutations that increased bradykinin activity and cause a decreased threshold for activation of the plasma contact system thus leading to the symptoms of angioedema.[6]
Hereditary angioedema with normal C1-inhibitor is a genetically heterogeneous disorder. Several molecular subtypes have been identified. A first subtype, identified in 2006, is caused by mutations of the F12 gene encoding coagulation factor XII (also known as Hageman factor).[23][24] All four mutations known so far, the two originally described missense mutations in exon 9 as well as the two additional, very rare mutations described later, affect the proline-rich region of coagulation factor XII.[23][24][21] An accelerated activation of the kallikrein-kinin system appears to represent the pathomechanism through that the F12 mutations cause angioedema.[25]
Clinical manifestation of hereditary angioedema due to a F12 mutation occurs preferably, but not exclusively in female mutation carriers. The remarkable estrogen-sensitivity is a characteristic feature of the HAE type caused by a F12 mutation. For example, estrogen-containing oral contraceptives play an important role in triggering angioedema attacks. Exacerbation of symptoms during pregnancy is also a common observation.[26][22] Hereditary angioedema due to Factor XII dysfunction is the most common subtype of type III angioedema.[6]
A second molecular subtype of HAE with normal C1-inhibitor is caused by a mutation of the plasminogen gene, namely a rare missense mutation within the kringle 3 domain of plasminogen, resulting in a novel type of dysplasminogenemia.[27] The mutation creates a new lysine-binding site within the kringle 3 domain and alters the glycosylation of plasminogen.[27] The mutant plasminogen protein has been shown to be a highly efficient kininogenase that directly releases bradykinin from high- and low-molecular-weight kininogen.[28] Tongue swellings are a very frequent and characterizing symptom in patients with hereditary angioedema due to a plasminogen mutation.[27][22]
Very rare observations have suggested that mutations of the following genes may also be responsible for the development of hereditary angioedema with normal C1-inhibitor: angiopoietin 1 [HAE with a angiopoietin 1 (ANGPT1) mutation]; myoferlin [HAE with a myoferlin (MYOF) mutation]; kininogen 1 [HAE with a kininogen 1 (KNG1) mutation]; and heparan sulfate-glucosamine 3-sulfotransferase 6 [HAE with a heparan sulfate-glucosamine 3-sulfotransferase 6 (HS3ST6) mutation].[24][22]
However, for a large proportion of cases with hereditary angioedema with normal C1-inhibitor the genetic cause remains unknown.[6]
Prevention
Treatment with ACE inhibitors is contraindicated in this condition, as these drugs can lead to bradykinin accumulation, which can precipitate disease episodes.[29][30]
Long-term
People in whom episodes occur at least once a month or who are at high risk of developing laryngeal edema require long-term prevention. There are several phase III clinical trials addressing HAE prophylaxis and therapy. These have led to the licensing of pdC1INH in many parts of the world; bradykinin receptor antagonists (icatibant) in Europe; kallikrein inhibitors (ecallantide and lanadelumab) in the United States; and recombinant C1-INH replacement therapy (rhC1INH; conestat alfa) in Europe. Tranexamic acid has been shown to be relatively ineffective therapy. Danazol prophylaxis remains an option but therapeutic agents are now being used more for prophylaxis because of danazol's adverse events.[31] For people requiring long-term prophylaxis, home therapy which allows people to self-administer the product, is considered an integral part of allowing patients a normal quality of life.
In 2018, the U.S. Food and Drug Administration (FDA) approved lanadelumab, an injectable monoclonal antibody, to prevent attacks of HAE types I and II in people over age 12. Lanadelumab inhibits the plasma enzyme kallikrein, which liberates the kinins bradykinin and kallidin from their kininogen precursors and is produced in excess in individuals with HAE types I and II.[32][33]
Berotralstat was approved in the US in December 2020, for the prevention of attacks of hereditary angioedema in people over twelve years of age.[34][35]
Short-term
Short-term prevention is normally administered before surgery or dental treatment. In Germany, C1-INH concentrate is used for this and given 1–1.5 hours before the procedure. In countries where C1-inhibitor concentrate is not available or only available in an emergency (laryngeal edema), high-dose androgen treatment is administered for 5–7 days.
Management
The aim of acute treatment is to halt progression of the edema as quickly as possible, which can be life-saving, particularly if the swelling is in the larynx. In Germany, most acute treatment consists of C1 inhibitor concentrate from donor blood, which must be administered intravenously; however, in most European countries, C1 inhibitor concentrate is only available to patients who are participating in special programs. In emergency situations where C1 inhibitor concentrate is not available, fresh frozen plasma (FFP) can be used as an alternative, as it also contains C1 inhibitor.
Other treatment modalities can stimulate the synthesis of C1 inhibitor, or reduce C1 inhibitor consumption. Purified C1 inhibitor, derived from human blood, has been used in Europe since 1979. Several C1 inhibitor treatments are now available in the U.S. Food and Drug Administration and two C1 inhibitor products are now available in Canada. Berinert P (CSL Behring), which is pasteurized, was approved by the F.D.A. in 2009 for acute attacks. Cinryze (ViroPharma), which is nanofiltered, was approved by the F.D.A. in 2008 for prophylaxis. Ruconest (Pharming) is a recombinant C1 inhibitor approved in the US and Europe that does not carry the risk of infectious disease transmission due to human blood-borne pathogens.[36]
The medication ecallantide inhibits plasma kallikrein and was approved by the F.D.A. (but not in Europe) for acute attacks in 2009. Icatibant inhibits the bradykinin B2 receptor, and was approved in Europe and the USA.[36][37] In HAE, specific stimuli that have previously led to attacks may need to be avoided in the future. It does not respond to antihistamines, corticosteroids, or epinephrine.
In February 2023, the FDA approved the expanded use of lanadelumab (Takhzyro) to prevent attacks of hereditary angioedema in children aged 2 to 12 years of age.[38]
Prognosis
About 25% of those affected die in the first two decades of life, mainly due to lack of treatment.[39]
Epidemiology
Data regarding the epidemiology of angioedema is limited. The incidence of HAE is one in 10,000–50,000 people in the United States and Canada. Mortality rates are estimated at 15–33%, resulting primarily from laryngeal edema and asphyxiation. HAE leads to 15,000–30,000 emergency department visits per year.[40][41]
Society and culture
There are national associations for HAE patients and their families in a number of countries around the world. These national associations are members of the global organization HAEi - International Patient Organization for C1-Inhibitor Deficiencies. HAEi is dedicated to raising awareness of C1 inhibitor deficiencies around the world. It is a non-profit international network established to promote co-operation, co-ordination and information sharing between HAE specialists and national HAE patient associations in order to help facilitate the availability of effective diagnosis and management of C1 inhibitor deficiencies throughout the world.[42]
Each year on 16 May, HAEi and the HAE community raise awareness of HAE with the international hae day :-)[43][44][45]
The Assistance Fund Inc. is an American nonprofit organization that offers co-pay assistance for medications that treat HAE and is open to any American Citizens or landed immigrants who have insurance.
A column on puzzling medical cases in The New York Times Magazine featured a 45-year-old woman with intestinal swelling who was ultimately diagnosed as having type III HAE. The patient’s HAE attack had been triggered by exposure to estrogen from her oral contraceptive.[46]
Research
Clinical development of several new active substances, which intervene in the disease process in different ways, is currently ongoing.
Pharming Group NV announced on 24 June 2010 that the European Medicines Agency has adopted a positive opinion on conestat alfa (trade name Ruconest), a C1-inhibitor for the treatment of acute angioedema attacks.[47]
Ecallantide, a peptide inhibitor of kallikrein, has received orphan status for HAE and has shown positive results in phase III trials.[48]
Icatibant (marketed as Firazyr) is a selective bradykinin receptor antagonist, which has been approved in Europe and was approved in the US by the FDA in Aug 2011.[49] After initial borderline results this drug was shown to be effective in phase III trials.[50] Cinryze has been approved by the FDA in October 2008.[51]
References
- 1 2 3 4 5 6 7 8 9 "Hereditary angioedema". GARD. 2017. Archived from the original on 4 July 2017. Retrieved 10 July 2017.
- 1 2 3 4 5 6 7 8 "Orphanet: Hereditary angioedema". www.orpha.net. August 2011. Archived from the original on 9 October 2015. Retrieved 10 July 2017.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Reference, Genetics Home (5 July 2017). "hereditary angioedema". Genetics Home Reference. Archived from the original on 10 July 2017. Retrieved 10 July 2017.
- ↑ "Hereditary Angioedema - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). 2008. Archived from the original on 14 July 2017. Retrieved 10 July 2017.
- ↑ Levin, Alex V.; Enzenauer, Robert W. (2017). The Eye in Pediatric Systemic Disease. Springer. p. 71. ISBN 9783319183893. Archived from the original on 2017-09-10.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Busse PJ, Christiansen SC (2020). "Hereditary Angioedema". N Engl J Med. 382 (12): 1136–1148. doi:10.1056/NEJMra1808012. PMID 32187470. S2CID 214584080.
- 1 2 Nzeako UC, Frigas E, Tremaine WJ (November 2001). "Hereditary angioedema: a broad review for clinicians". Arch Intern Med. 161 (20): 2417–29. doi:10.1001/archinte.161.20.2417. PMID 11700154.
- ↑ Ferraro MF, Moreno AS, Castelli EC, Donadi EA, Palma MS, Arcuri HA, Lange AP, Bork K, Sarti W, Arruda LK (October 2011). "A single nucleotide deletion at the C1 inhibitor gene as the cause of hereditary angioedema: insights from a Brazilian family". Allergy. 66 (10): 1384–90. doi:10.1111/j.1398-9995.2011.02658.x. PMID 21623829. S2CID 23036731.
- ↑ Bafunno V, Bova M, Loffredo S, Divella C, Petraroli A, Marone G, Montinaro V, Margaglione M, Triggiani M (March 2014). "Mutational spectrum of the c1 inhibitor gene in a cohort of Italian patients with hereditary angioedema: description of nine novel mutations". Ann Hum Genet. 78 (2): 73–82. doi:10.1111/ahg.12052. PMID 24456027.
- ↑ Weiler CR, van Dellen RG (July 2006). "Genetic test indications and interpretations in patients with hereditary angioedema". Mayo Clin Proc. 81 (7): 958–72. doi:10.4065/81.7.958. PMID 16835976.
- ↑ "Inheritance of Hereditary Angioedema - Patient Information - Shire's Brave Community". Archived from the original on 2014-05-06. Retrieved 2014-05-05.
- ↑ "Management of angioedema in the ER" (PDF). Archived from the original (PDF) on 2014-11-03. Retrieved 2014-11-03.
- ↑ Zingale LC, Beltrami L, Zanichelli A, et al. (October 2006). "Angioedema without urticaria: a large clinical survey". CMAJ. 175 (9): 1065–70. doi:10.1503/cmaj.060535. PMC 1609157. PMID 17060655.
- ↑ "Diagnostic Algorithm". HAE Canada. Archived from the original on 2014-11-03. Retrieved 2014-11-03.
- ↑ Donaldson VH, Evans RR (July 1963). "A biochemical abnormality in hereditary angioneurotic edema: Absence of serum inhibitor of C' 1-esterase". Am. J. Med. The American Journal of Medicine. 35 (1): 37–44. doi:10.1016/0002-9343(63)90162-1. PMID 14046003.
- ↑ Rosen FS, Pensky J, Donaldson V, Charache P (May 14, 1965). "Hereditary angioneurotic edema: Two genetic variants". Science. 148 (3672): 957–958. Bibcode:1965Sci...148..957R. doi:10.1126/science.148.3672.957. PMID 14277836. S2CID 32776518.
- 1 2 Carugati A, Pappalardo E, Zingale LC, Cicardi M (August 2001). "C1-inhibitor deficiency and angioedema". Mol. Immunol. Molecular Immunology. 38 (2–3): 161–173. doi:10.1016/s0161-5890(01)00040-2. PMID 11532278.
- 1 2 Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, López-Lera A, López-Trascasa M, Tosi M, Drouet C (January 2020). "SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes". Hum. Mutat. Human Mutation. 41 (1): 38–57. doi:10.1002/humu.23917. PMID 31517426.
- ↑ Pappalardo E, Cicardi M, Duponchel C, Carugati A, Choquet S, Agostoni A, Tosi M (December 2000). "Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema". J. Allergy Clin. Immunol. The Journal of Allergy and Clinical Immunology. 106 (6): 1147–1154. doi:10.1067/mai.2000.110471. PMID 11112899.
- ↑ Bork K, Barnstedt SE, Koch P, Traupe H (July 15, 2000). "Hereditary angioedema with normal C1-inhibitor activity in women". Lancet. The Lancet. 356 (9225): 213–217. doi:10.1016/S0140-6736(00)02483-1. PMID 10963200. S2CID 30105665.
- 1 2 Banday AZ, Kaur A, Jindal AK, Rawat A, Singh S (March 2020). "An update on the genetics and pathogenesis of hereditary angioedema". Genes Dis. Genes & Diseases. 7 (1): 75–83. doi:10.1016/j.gendis.2019.07.002. PMC 7063419. PMID 32181278.
- 1 2 3 4 Jones D, Zafra H, Anderson J (April 22, 2023). "Managing Diagnosis, Treatment, and Burden of Disease in Hereditary Angioedema Patients with Normal C1-Esterase Inhibitor". J Asthma Allergy. Journal of Asthma and Allergy. 16: 447–460. doi:10.2147/JAA.S398333. PMC 10132308. PMID 37124440.
- 1 2 Dewald G, Bork K (May 19, 2006). "Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor". Biochem. Biophys. Res. Commun. Biochemical-and-Biophysical-Research-Communications. 343 (4): 1286–1289. doi:10.1016/j.bbrc.2006.03.092. PMID 16638441.
- 1 2 3 Santacroce R, D'Andrea G, Maffione AB, Margaglione M, d'Apolito M (May 9, 2021). "The Genetics of Hereditary Angioedema: A Review". J. Clin. Med. Journal of Clinical Medicine. 10 (9): 2023. doi:10.3390/jcm10092023. PMC 8125999. PMID 34065094.
- ↑ Shamanaev A, Dickeson SK, Ivanov I, Litvak M, Sun MF, Kumar S, Cheng Q, Srivastava P, He TZ, Gailani D (May 23, 2023). "Mechanisms involved in hereditary angioedema with normal C1-inhibitor activity". Front. Physiol. Frontiers in Physiology. 14: 1146834. doi:10.3389/fphys.2023.1146834. PMC 10242079. PMID 37288434.
- ↑ Deroux A, Boccon-Gibod I, Fain O, Pralong P, Ollivier Y, Pagnier A, Djenouhat K, Du-Thanh A, Gompel A, Faisant C, Launay D, Bouillet L (September 2016). "Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema". Clin. Exp. Immunol. Clinical & Experimental Immunology. 185 (3): 332–337. doi:10.1111/cei.12820. PMC 4991515. PMID 27271546.
- 1 2 3 Dewald G (March 25, 2018). "A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor". Biochem Biophys Res Commun. Biochemical-and-Biophysical-Research-Communications. 498 (1): 193–198. doi:10.1016/j.bbrc.2017.12.060. PMID 29548426.
- ↑ Dickeson SK, Kumar S, Sun MF, Mohammed BM, Phillips DR, Whisstock JC, Quek AJ, Feener EP, Law RHP, Gailani D (May 5, 2022). "A mechanism for hereditary angioedema caused by a lysine 311-to-glutamic acid substitution in plasminogen". Blood. Blood. 139 (18): 2816–2829. doi:10.1182/blood.2021012945. PMC 9074402. PMID 35100351.
- ↑ Dendorfer A, Wolfrum S, Wagemann M, Qadri F, Dominiak P (May 2001). "Pathways of bradykinin degradation in blood and plasma of normotensive and hypertensive rats". Am. J. Physiol. Heart Circ. Physiol. 280 (5): H2182–8. doi:10.1152/ajpheart.2001.280.5.H2182. PMID 11299220. S2CID 5898184.
- ↑ Kuoppala A, Lindstedt KA, Saarinen J, Kovanen PT, Kokkonen JO (April 2000). "Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma". Am. J. Physiol. Heart Circ. Physiol. 278 (4): H1069–74. doi:10.1152/ajpheart.2000.278.4.H1069. PMID 10749699. S2CID 10371501.
- ↑ Hereditary angioedema: beyond international consensus - circa December 2010 - The Canadian Society of Allergy and Clinical Immunology Dr. David McCourtie Lecture
- ↑ "FDA Approves Takhzyro (lanadelumab-flyo) for Hereditary Angioedema". Drugs.com.
- ↑ "FDA OKs New Prophylactic Drug for Rare Hereditary Angioedema". Medscape.
- ↑ "Drug Trials Snapshot: Orladeyo". U.S. Food and Drug Administration. 3 December 2020. Retrieved 25 December 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ↑ "Orladeyo: FDA-Approved Drugs". U.S. Food and Drug Administration (FDA). Retrieved 25 December 2020.
- 1 2 Morgan
- ↑ Firazyr [package insert]. Lexington, MA: Shire Orphan Therapies, Inc; 2011.
- ↑ "FDA Approves sBLA for Takeda's Takhzyro for HAE Attacks in 2y/o and up". Takeda (Press release). Retrieved 2023-02-06.
- ↑ Varga, Lilian; Farkas, Henriette (2008-11-01). "Treatment of type I and II hereditary angioedema with Rhucin, a recombinant human C1 inhibitor". Expert Review of Clinical Immunology. 4 (6): 653–661. doi:10.1586/1744666X.4.6.653. ISSN 1744-666X. PMID 20477114. S2CID 11656834.
- ↑ From the: Pinnacle Health System, Harrisburg Hospital, Department of Internal Medicine, 111 South Front Street, Harrisburg, PA 17101, Update on treatment for her
- ↑ "Update on treatment of hereditary angioedema" Buyantseva, Larisa, Sardana, Niti and Craig, Timothy
- ↑ "HAEi website". Archived from the original on 2017-09-08. Retrieved 2017-09-10.
- ↑ Chapman, Mary (5 May 2022). "HAE Awareness Day 'Stepping Up' With Activity Challenges, Storytelling". Retrieved 2022-05-23.
- ↑ Inouye, Daniel K. (2012-01-31). "S.Res.286 - 112th Congress (2011-2012): A resolution recognizing May 16, 2012, as Hereditary Angioedema Awareness Day and expressing the sense of the Senate that more research and treatments are needed for Hereditary Angioedema". www.congress.gov. Retrieved 2022-05-23.
- ↑ Jindal, A. K.; Bishnoi, A.; Dogra, S. (2021). "Hereditary Angioedema: Diagnostic Algorithm and Current Treatment Concepts". Indian Dermatology Online Journal. 12 (6): 796–804. doi:10.4103/idoj.idoj_398_21. PMC 8653746. PMID 34934714.
- ↑ Sanders, Lisa (28 December 2020). "The New York Times Magazine". The New York Times. Retrieved 2020-12-29.
- ↑ Pharming: Pharming Receives Positive Opinion From European Medicines Agency On Rhucin Product name in Europe changed to Ruconest
- ↑ Lehmann A (August 2008). "Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery". Expert Opin Biol Ther. 8 (8): 1187–99. doi:10.1517/14712598.8.8.1187. PMID 18613770. S2CID 72623604.
- ↑ Jerini AG (2008-07-15). "Jerini Receives European Commission Approval for Firazyr (Icatibant) in the Treatment of HAE - Press release". Archived from the original on 2018-09-29. Retrieved 2008-07-28.
- ↑ Bernstein JA (January 2008). "Hereditary angioedema: a current state-of-the-art review, VIII: current status of emerging therapies". Ann. Allergy Asthma Immunol. 100 (1 Suppl 2): S41–6. doi:10.1016/S1081-1206(10)60585-6. PMID 18220151.
- ↑ Reuters: UPDATE 1-US clears Lev Pharma drug for rare swelling disease Archived 2009-09-09 at the Wayback Machine
{kind=link}
{kind=link}
Further reading
External links
 Media related to Hereditary angioedema at Wikimedia Commons
Media related to Hereditary angioedema at Wikimedia Commons- Hereditary angioedema at Curlie