| Catecholaminergic polymorphic ventricular tachycardia | |
|---|---|
| Other names | CPVT |
.jpg.webp) | |
| Bidirectional ventricular tachycardia in a patient with CPVT | |
| Specialty | Cardiology |
| Symptoms | Blackouts, sudden cardiac death[1] |
| Usual onset | Childhood / adolescence |
| Causes | Genetic |
| Risk factors | Family history |
| Diagnostic method | Electrocardiogram (ECG), genetic testing, adrenaline provocation, exercise testing[1] |
| Differential diagnosis | Long QT syndrome, Brugada syndrome, Andersen-Tawil syndrome, Early repolarization syndrome |
| Treatment | Avoidance of strenuous exercise, medication, implantable cardioverter defibrillator[2] |
| Medication | Beta-adrenoceptor blockers, Verapamil, Flecainide[2] |
| Prognosis | 13–20% life threatening arrhythmias over 7–8 years[3] |
| Frequency | 1:10,000[4] |
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited genetic disorder that predisposes those affected to potentially life-threatening abnormal heart rhythms or arrhythmias. The arrhythmias seen in CPVT typically occur during exercise or at times of emotional stress, and classically take the form of bidirectional ventricular tachycardia or ventricular fibrillation. Those affected may be asymptomatic, but they may also experience blackouts or even sudden cardiac death.
CPVT is caused by genetic mutations affecting proteins that regulate the concentrations of calcium within cardiac muscle cells. The most commonly identified gene is RYR2, which encodes a protein included in an ion channel known as the ryanodine receptor; this channel releases calcium from a cell's internal calcium store, the sarcoplasmic reticulum, during every heartbeat.
CPVT is often diagnosed from an ECG recorded during an exercise tolerance test, but it may also be diagnosed with a genetic test. The condition is treated with medication including beta-adrenoceptor blockers or flecainide, or with surgical procedures including sympathetic denervation and implantation of a defibrillator. It is thought to affect as many as one in ten thousand people and is estimated to cause 15% of all unexplained sudden cardiac deaths in young people. The condition was first defined in 1978,[5] and the underlying genetics were described in 2001.[6]
Signs and symptoms
.png.webp)
Although individuals with CPVT may not experience any symptoms, the most commonly reported symptoms are blackouts or sudden loss of consciousness, referred to as syncope.[1] These blackouts often occur during exercise or as a response to emotional stress—situations in which chemical messengers known as catecholamines, such as adrenaline, are released within the body. Blackouts may be misinterpreted as being caused by simple faints or epilepsy, often leading to delays in reaching the correct diagnosis.[7] In a third of those affected, the first manifestation of the disease may be cardiac arrest, potentially leading to sudden death.[8] This can occur in very young children, presenting as sudden infant death syndrome or 'cot death'.[1] Approximately 30% of those with CPVT will have a family member who has experienced blackouts, seizures, or sudden death in response to exercise or stress.[7]
In those with CPVT, catecholamine release can lead to an abnormal heart rhythm or arrhythmia known as ventricular tachycardia.[4] The ventricular tachycardia may take a characteristic form known as bidirectional ventricular tachycardia. This form of ventricular tachycardia occurs relatively infrequently, but if seen is suggestive of an underlying diagnosis of CPVT or the related condition Andersen-Tawil syndrome.[8][9] These ventricular arrhythmias in some cases terminate by themselves, causing a blackout from which the person then recovers. However, if the abnormal heart rhythm continues, it can degenerate into a more dangerous arrhythmia known as ventricular fibrillation causing a cardiac arrest and, if untreated, sudden death.[10]
There are typically very few abnormal signs on clinical examination in persons with CPVT. However, those with CPVT may develop a less serious heart rhythm disturbance called atrial fibrillation, which can be detected on examination as an irregular pulse.[8] Furthermore, approximately 20% of those with CPVT have a slow resting heart rate known as a sinus bradycardia.[4]
Mechanism
Excitation-contraction coupling
The arrhythmias that those with CPVT experience are caused by abnormalities in the way that cardiac muscle cells control their levels of calcium.[11] Calcium interacts with the protein fibres or myofibrils inside the cell that allow the cell to contract, and the concentration of calcium within each cell needs to be tightly regulated. During each heartbeat, the concentration of calcium must rise to allow the muscle to contract and then fall to allow the muscle to relax, a process achieved by using a store within the cell known as the sarcoplasmic reticulum.[12]
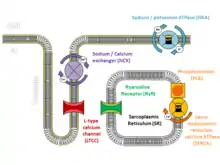
At the start of each heartbeat, calcium is released from the sarcoplasmic reticulum through specialised channels known as ryanodine receptors.[12] Ryanodine receptors open when the concentration of calcium near the channel increases. This happens when, in response to an electrical signal from the cell membrane called an action potential, a small amount of calcium flows across the cell membrane into the cell through L-type calcium channels, many of which are located on specialised inpouchings of the membrane called T-tubules designed to bring these surface ion channels close to the sarcoplasmic reticulum.[13]
The increase in calcium concentration triggers ryanodine receptors on the sarcoplasmic reticulum to release a puff of calcium known as a calcium spark. Each spark triggers the release of further sparks from neighbouring ryanodine receptors to create an organised rise of calcium throughout the cell known as a calcium transient. At the end of each heartbeat, calcium is pumped back by a protein called SERCA along with its regulatory protein phospholamban. The calcium is then held within the sarcoplasmic reticulum by a protein called calsequestrin.[12]
Fine-tuning of this process can be achieved by phosphorylating these proteins. As an example, during exercise catecholamines activate beta-adrenoceptors on the cell surface, which trigger protein kinase A to phosphorylate the L-type calcium channel, increasing the flow of calcium into the cell. Simultaneously, phosphorylation of the regulatory protein phospholamban causes more calcium to be drawn up into the sarcoplasmic reticulum. The overall effect of this is to generate a larger calcium transient with each beat, leading to a more forceful contraction.[13]
Calcium-dependent arrhythmias
Alterations to the proteins involved in excitation-contraction coupling can disrupt this carefully regulated process. In those with CPVT, the normally tight regulation of calcium can become deranged, leading to arrhythmias.[11] While calcium is generally released from the sarcoplasmic reticulum in response to an action potential, calcium sparks can also occur spontaneously. In a healthy heart, a spontaneous calcium spark is generally an isolated event and goes no further, but if ryanodine receptors or the proteins that regulate them are abnormal, these sparks can trigger releases from neighbouring ryanodine receptors which spread throughout the cell as a calcium wave.[11] These calcium waves are much more likely to occur when cardiac muscle cells are stimulated by catecholamines such as adrenaline, which increase the concentration of calcium within the sarcoplasmic reticulum and sensitise the ryanodine receptors.[14] The uncontrolled wave of calcium can be forced out through the cell membrane via the sodium-calcium exchanger, causing an electric current known as a delayed afterdepolarisation. Afterdepolarisations, if large enough, can trigger additional action potentials, premature ventricular contractions, or sustained arrhythmias.[15]
Causes
| Type | OMIM | Gene | Locus | Inheritance | Notes |
|---|---|---|---|---|---|
| CPVT1 | 604772 | RYR2 | 1q42.1-q43 | AD | Ryanodine receptor—releases calcium from the sarcoplasmic reticulum |
| CPVT2 | 611938 | CASQ2 | 1p13.3-p11 | AR | Calsequestrin—calcium buffer within the sarcoplasmic reticulum[8] |
| CPVT3 | 614021 | TECRL | 7p22-p14 | AR | Trans-2,3-enoyl-CoA reductase-like protein—interacts with ryanodine receptors and calsequestrin |
| CPVT4 | 614916 | CALM1 | 14q32.11 | AD | Calmodulin—stabilises ryanodine receptors |
| CPVT5 | 615441 | TRDN | 6q22.31 | AR | Triadin—forms complex with calsequestrin to interact with ryanodine receptors |
CPVT can be caused by mutations in several genes, all of which are responsible for regulating the concentrations of calcium within cardiac muscle cells. The most commonly identified genetic mutation in CPVT is a mutation in the RYR2 gene that encodes the cardiac ryanodine receptor, responsible for releasing calcium from the sarcoplasmic reticulum.[16] Mutations associated with CPVT have also been identified in the CASQ2 gene which encodes calsequestrin,[17] a protein that binds calcium within the sarcoplasmic reticulum. Other genes associated with rarer, atypical forms of CPVT[16] include TECRL encoding Trans-2,3-enoyl-CoA reductase-like protein, CALM1, CALM2 and CALM3 all encoding Calmodulin, and TRDN encoding Triadin.[18]
CPVT1: RYR2 mutations
The most commonly identified genetic mutations in those with CPVT occur in the RYR2 gene which encodes the cardiac ryanodine receptor.[11] Mutations in this gene lead to an autosomal dominant form of typical CPVT known as CPVT1.[16] While the precise effect differs between specific mutations in this gene, many RYR2 mutations cause the ryanodine receptor to open in response to lower concentrations of calcium—the threshold for calcium release is lower.[11] As a result, the sarcoplasmic reticulum spontaneously releases calcium through these abnormal ryanodine receptors when the concentration of calcium within the sarcoplasmic reticulum rises, a process known as store-overload induced calcium release.[10] Sarcoplasmic reticulum calcium content increases in response to stimulation from catecholamines, explaining why arrhythmias in those with CPVT occur at times when catecholamine levels are elevated. Some suggest that the increased sensitivity to calcium occurs only when the ryanodine receptor is phosphorylated by protein kinase A, while other suggest that the increased sensitivity also occurs under resting conditions.[11]
Two theories have been proposed for the underlying mechanism by which mutations in RYR2 promote store-overload induced calcium release: domain unzipping and FKBP12.6 unbinding.[10] Domain unzipping refers to the separation of two important regions of the ryanodine receptor, the N-terminus and the central domain. Through this mechanism, a mutation might destabilise the ryanodine receptor's closed state and increase its sensitivity to calcium.[10] A second potential mechanism involves the regulatory protein FKBP12.6, a protein that binds to and stabilises the ryanodine receptor. The binding of FKBP12.6 to the ryanodine receptor is regulated by phosphorylation. Phosphorylation by protein kinase A leads to the dissociation of FKBP12.6, rendering the ryanodine receptor more sensitive to cytosolic calcium. RYR2 mutations may interfere with the binding of FKB12.6 to the ryanodine receptor and thereby increase sensitivity to calcium. It is likely that FKBP12.6 plays a role in some CPVT mutations but not others.[10]
RYR2 mutations responsible for CPVT are mainly found in four major domains of the gene.[10] Mutations affecting domains III and IV of the gene (corresponding to the N-terminal region of the protein and cytosolic linker respectively) occur in 46% of cases. Mutations are seen less frequently affecting domains I and II, both of which encode sections of the N-terminal region of the protein.[10] RYR2 mutations associated with CPVT that occur outside these four domains are very rare, being responsible for as few as 10% of reported cases. RYR2 mutations are most often missense mutations, such as single nucleotide substitutions causing one amino acid to be replaced by another, although in-frame substitutions and duplications have been described. More damaging nonsense mutations have not been reported in association with CPVT, potentially because these variants may lead to different cardiac diseases such as cardiomyopathies.[10]
CPVT2: CASQ2 mutations
Mutations in the CASQ2 gene are associated with an autosomal recessive form of typical CPVT known as CPVT2.[16] This gene encodes calsequestrin, the major calcium-binding protein and calcium buffer within the sarcoplasmic reticulum.[19][20] Mutations in CASQ2 account for only 3-5% of cases of CPVT.[16] Fourteen mutations in CASQ2 have been identified in association with CPVT. Two of these are nonsense mutations causing the protein to be abnormally short, and two are deletion mutations, while ten are missense mutations that substitute one amino acid for another in the chain forming the protein.[11]
Mutations in CASQ2 cause a decrease in sarcoplasmic reticulum calcium-buffering capacity. This means that abrupt changes in sarcoplasmic total calcium will be buffered less and therefore translate to larger shifts in free calcium. The higher peaks in free calcium have greater potential to cause store-overload induced calcium release from the sarcoplasmic reticulum, leading to afterdepolarisations.[10]
In addition to its role as a calcium buffer, calsequestrin also regulates the release of calcium from the sarcoplasmic reticulum by directly modulating ryanodine receptors. When the concentration of calcium is low, calsequestrin monomers form a complex with the proteins triadin and junctin, which inhibit ryanodine receptors.[19] However, at high calcium concentrations, calsequestrin forms polymers that dissociate from the ryanodine receptor channel complex, removing the inhibitory response and increasing the sensitivity of the ryanodine receptor to spontaneously releasing calcium.[10]
Decreased CASQ2 is also associated with high levels of calreticulin, a protein which among other roles regulates the reuptake of calcium into the sarcoplasmic reticulum by SERCA.[21] In the absence of CASQ2, calreticulin levels increase and provide some compensatory calcium binding within the sarcoplasmic reticulum. It is possible that calreticulin may contribute to the generation of arrhythmias seen in association with CASQ2 mutations.[21]
Diagnosis
.jpg.webp)
CPVT may be challenging to diagnose as the structure of the heart appears normal in those affected by the condition when assessed using an echocardiogram, cardiac MRI scan or cardiac CT scan, while the electrical function of the heart also appears normal at rest when assessed using a standard 12-lead ECG.[1][2] However, in response to exercise or catecholamines such as adrenaline, abnormal heart rhythms such as bidirectional ventricular tachycardia or frequent polymorphic ventricular ectopic beats may be seen.[22]
12-lead ECG
The resting 12-lead ECG is a useful test to differentiate CPVT from other electrical diseases of the heart that can cause similar abnormal heart rhythms. Unlike conditions such as long QT syndrome and Brugada syndrome, the resting 12-lead ECG in those with CPVT is generally normal.[8] However, approximately 20% of those affected have a slow resting heart rate or sinus bradycardia.[8]
Exercise and other provocative testing

Exercise testing, commonly performed on a treadmill or stationary bicycle, can help to diagnose CPVT. During the test, those with CPVT often experience ectopic beats, which may progress to bidirectional and then polymorphic ventricular tachycardia as the intensity of exercise increases.[23] Some of those suspected of having CPVT, such as young children, may not be able to perform an exercise tolerance test. In these cases, alternative forms of testing include adrenaline provocation testing, during which adrenaline is infused into a vein at gradually increasing doses under close supervision and ECG monitoring.[22] Additionally, long term or Holter ECG monitoring can be performed, although this form of testing is less likely to detect an arrhythmia. Invasive electrophysiological studies do not provide useful information to help diagnose CPVT or to assess the risk of life-threatening arrhythmias.[2][22]
Genetic testing
CPVT can also be diagnosed by identifying a disease-causing mutation in a gene associated with CPVT using genetic testing.[2][22] This technique may be the only way to identify the condition in someone suspected of having CPVT who has died, and in this case may be known as a molecular autopsy.[24]
Treatment
Treatments for CPVT aim to prevent lethal abnormal heart rhythms from occurring, and to rapidly restore a normal rhythm if they do occur. As the arrhythmias in CPVT generally occur at times when the heart is exposed to high levels of adrenaline or other similar chemical messengers (catecholamines), many treatments for CPVT aim to lower the levels of catecholamines the heart is exposed to or block their effects on the heart.[8]
The first-line treatment for those with CPVT involves lifestyle advice. This includes avoiding competitive sports, very strenuous exercise and highly stressful environments, as high levels of adrenaline can occur in these settings, which can provoke arrhythmias.[2]
Medication
Several medications can be useful for those with CPVT. The mainstays of treatment are beta blockers, which block the effects of adrenaline and other catecholamines on the heart, reducing the chance of abnormal heart rhythms developing.[2] Of all the beta blockers, nadolol has been proven to be the most effective for treating CPVT.[25] This drug lowers the heart rate to a greater extent than other beta blockers and only needs to be taken once daily, reducing the risk of missed doses. Nadolol may be difficult to obtain and is not available in all countries, and an alternative beta blocker suitable for use in CPVT may be propranolol, which however has a more complex dosing regimen.[25] Recently published data suggest that the use of selective beta blockers, such as atenolol, bisoprolol, or metoprolol, is associated with very high treatment failure rates.[25]
Flecainide is a class 1c antiarrhythmic drug that is recommended for those with CPVT who experience abnormal heart rhythms despite taking a beta blocker.[2] Flecainide reduces the risk of arrhythmias in those with CPVT, but it remains uncertain how Flecainide achieves this. Some have suggested that Flecainide directly interacts with the cardiac ryanodine receptor, which is frequently abnormal in those with CPVT, while other suggest that the anti-arrhythmic effects of Flecainide rely entirely on its sodium channel blocking effects.[26]
Verapamil is a calcium channel antagonist that, when combined with a beta blocker, may reduce the risk of arrhythmias in patients with CPVT.[27] Propafenone is another antiarrhythmic that may reduce the risk of arrhythmias, potentially through direct effects on the ryanodine receptor.[26]
Sympathetic denervation
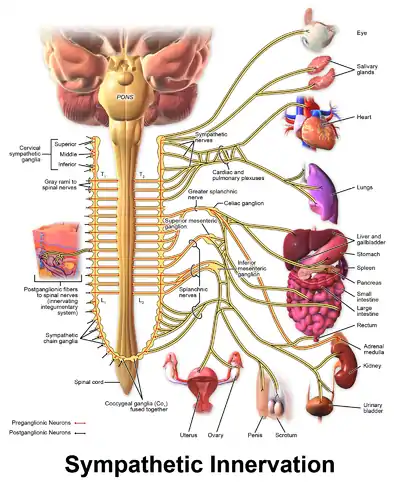
Some persons with CPVT continue to experience life-threatening arrhythmias despite pharmaceutical therapy. In this case a surgical procedure can be used to affect nerves supplying the heart that communicate using catecholamines.[2] A collection of nerves known as the sympathetic nervous system supply the heart as well as other organs. These nerves, when activated, encourage the heart to beat harder and faster.[28] The sympathetic nervous system uses noradrenaline, a catecholamine, as a chemical messenger or neurotransmitter, which can promote arrhythmias in those with CPVT.[2]
To prevent this, a region of the sympathetic nervous system can be intentionally damaged in an operation known as cardiac sympathetic denervation or sympathectomy.[29] While the sympathetic nervous system feeds into the heart from both sides, often only the left sided nerves are targeted during sympathectomy, although destruction of the nerves on both sides may be required.[29] Through this process, sympathectomy is effective at decreasing, but not abolishing, the risk of further life-threatening arrhythmias.[4]
Implantable cardioverter-defibrillator
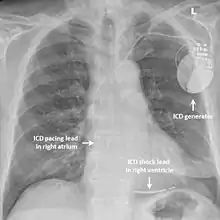
While medication and sympathectomy aim to prevent abnormal heart rhythms from occurring in the first place, an implantable defibrillator (ICD) may be used to treat arrhythmias that medication has failed to prevent and restore a normal heart rhythm.[2] These devices, usually implanted under the skin at the front of the chest below the shoulder, can continuously monitor the heart for abnormal heart rhythms. If a life-threatening arrhythmia is detected, the device can deliver a small electric shock to terminate the abnormal rhythm and restart the heart.[1]
Implantable defibrillators are often recommended for those with CPVT who have experienced blackouts, ventricular arrhythmias or cardiac arrest despite taking appropriate medication.[2] These devices are life-saving, as it has been shown that their use confers a significant survival benefit in patients with CPVT.[25] It has been suggested that the resulting surge of adrenaline caused by the pain of an electric shock from the device could theoretically bring on a cycle of recurrent arrhythmias and shocks known as an electrical storm,[4] and therefore it is strongly recommended that those with an ICD implanted for CPVT take a beta blocker to dampen the effects of adrenaline.[4]
Prognosis
A significant proportion of those with CPVT will experience a life-threatening abnormal heart rhythm, with estimates of this risk ranging from 13 to 20% over the course of 7–8 years.[3] Life-threatening arrhythmias are more likely to occur if CPVT has been diagnosed in childhood, if a person with CPVT does not take beta blockers, and if arrhythmias occur on exercise testing despite taking beta blockers.[2]
During treatment with nadolol, the preferred beta blocker for the treatment of CPVT, event rates have been estimated to be 0.8% per year. In patients treated with beta blockers, life-threatening arrhythmias are more likely if a person had already survived a cardiac arrest, had a syncope, or are carriers of disease-causing mutations affecting the highly conserved terminal portion of RYR2 gene,[25] called the C-terminal domain (amino acids 4889–4969).[30]
Epidemiology
CPVT is estimated to affect 1 in 10,000 people.[4] Symptoms from CPVT are typically first seen in the first or second decade of life,[25] and more than 60% of affected individuals experience their first episode of syncope or cardiac arrest by age 20.[1] Syncope during exercise or strong emotion should be considered a red flag,[25] as it is a characteristic of the disease. Lastly, a small number of patients may present later in life, and genetic testing in these patients frequently fails to identify a causative gene.[4]
History
In 1960, Norwegian cardiologist Knut Berg published a report on three sisters who had blackouts during exercise or emotional stress in what is now recognised as the first description of CPVT.[8] The bidirectional ventricular tachycardia associated with this condition was described in 1975.[1] The term "Catecholaminergic Polymorphic Ventricular Tachycardia" was first used in 1978.[5] In 1999, the first genetic mutation causing CPVT to be identified was localised to chromosome 1q42-q43,[31] which was found to be a variant in the RYR2 gene in 2001.[6] Ongoing research aims to identify better treatments for CPVT, to increase understanding of the mechanisms of arrhythmia, and to identify other genes causing the condition.[32]
References
- 1 2 3 4 5 6 7 8 Liu N, Napolitano C, Priori S (2013). Chapter 31: Catecholaminergic Polymorphic Ventricular Tachycardia. In: Electrical diseases of the heart. Volume 2, Diagnosis and treatment. Arthur Wilde, Ihor Gussak, Michael J. Ackerman, Win-Kuang Shen, and Charles Antzelevitch (2nd ed.). London: Springer. ISBN 978-1-4471-4978-1. OCLC 846445829.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ (November 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC)". European Heart Journal. 36 (41): 2793–2867. doi:10.1093/eurheartj/ehv316. hdl:11380/1140282. PMID 26320108.
- 1 2 Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, Klug D, Hayashi M, Takatsuki S, Villain E, Kamblock J, Messali A, Guicheney P, Lunardi J, Leenhardt A (May 2009). "Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia". Circulation. 119 (18): 2426–34. doi:10.1161/CIRCULATIONAHA.108.829267. PMID 19398665.
- 1 2 3 4 5 6 7 8 Obeyesekere MN, Antzelevitch C, Krahn AD (February 2015). "Management of ventricular arrhythmias in suspected channelopathies". Circulation: Arrhythmia and Electrophysiology. 8 (1): 221–31. doi:10.1161/CIRCEP.114.002321. PMID 25691556.
- 1 2 Coumel, Philippe; Fidelle, J; Lucet, V; Attuel, P; Bouvrain, Y (1978). "Catecholamine-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases". British Heart Journal.
- 1 2 Priori, Silvia G.; Napolitano, Carlo; Tiso, Natascia; Memmi, Mirella; Vignati, Gabriele; Bloise, Raffaella; Sorrentino, Vincenzo; Danieli, Gian Antonio (2001-01-16). "Mutations in the Cardiac Ryanodine Receptor Gene ( hRyR2 ) Underlie Catecholaminergic Polymorphic Ventricular Tachycardia". Circulation. 103 (2): 196–200. doi:10.1161/01.CIR.103.2.196. ISSN 0009-7322. PMID 11208676. S2CID 5872126.
- 1 2 Liu N, Colombi B, Raytcheva-Buono EV, Bloise R, Priori SG (May 2007). "Catecholaminergic polymorphic ventricular tachycardia". Herz. 32 (3): 212–7. doi:10.1007/s00059-007-2975-2. PMID 17497254. S2CID 40499394.
- 1 2 3 4 5 6 7 8 Lieve KV, van der Werf C, Wilde AA (May 2016). "Catecholaminergic Polymorphic Ventricular Tachycardia". Circulation Journal. 80 (6): 1285–91. doi:10.1253/circj.CJ-16-0326. PMID 27180891.
- ↑ Tristani-Firouzi M, Etheridge SP (2013). "Chapter 32 - Andersen-Tawil and Timothy Syndromes". In Gussak I, Antzelevitch C (eds.). Electrical diseases of the heart. Volume 1, Basic foundations and primary electrical diseases (2nd ed.). London: Springer. ISBN 978-1-4471-4881-4. OCLC 841465583.
- 1 2 3 4 5 6 7 8 9 10 Priori SG, Chen SR (April 2011). "Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis". Circulation Research. 108 (7): 871–83. doi:10.1161/CIRCRESAHA.110.226845. PMC 3085083. PMID 21454795.
- 1 2 3 4 5 6 7 Venetucci L, Denegri M, Napolitano C, Priori SG (October 2012). "Inherited calcium channelopathies in the pathophysiology of arrhythmias". Nature Reviews Cardiology. 9 (10): 561–75. doi:10.1038/nrcardio.2012.93. PMID 22733215. S2CID 24883043.
- 1 2 3 Bers, DM (2001). Excitation-contraction coupling and cardiac contractile force (2nd ed.). Dordrecht: Kluwer Academic Publishers. ISBN 978-0-7923-7157-1. OCLC 47659382.
- 1 2 Eisner DA, Caldwell JL, Kistamás K, Trafford AW (July 2017). "Calcium and Excitation-Contraction Coupling in the Heart". Circulation Research. 121 (2): 181–195. doi:10.1161/CIRCRESAHA.117.310230. PMC 5497788. PMID 28684623.
- ↑ Bers DM (January 2002). "Calcium and cardiac rhythms: physiological and pathophysiological". Circulation Research. 90 (1): 14–7. doi:10.1161/res.90.1.14. PMID 11786512.
- ↑ Pogwizd SM, Bers DM (February 2004). "Cellular basis of triggered arrhythmias in heart failure". Trends in Cardiovascular Medicine. 14 (2): 61–6. doi:10.1016/j.tcm.2003.12.002. PMID 15030791.
- 1 2 3 4 5 Priori, Silvia G.; Mazzanti, Andrea; Santiago, Demetrio J.; Kukavica, Deni; Trancuccio, Alessandro; Kovacic, Jason C. (May 2021). "Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia". Journal of the American College of Cardiology. 77 (20): 2592–2612. doi:10.1016/j.jacc.2020.12.073. PMID 34016269. S2CID 235072676.
- ↑ Lahat, Hadas; Pras, Elon; Olender, Tsviya; Avidan, Nili; Ben-Asher, Edna; Man, Orna; Levy-Nissenbaum, Etgar; Khoury, Asad; Lorber, Avraham; Goldman, Boleslaw; Lancet, Doron (December 2001). "A Missense Mutation in a Highly Conserved Region of CASQ2 Is Associated with Autosomal Recessive Catecholamine-Induced Polymorphic Ventricular Tachycardia in Bedouin Families from Israel". The American Journal of Human Genetics. 69 (6): 1378–1384. doi:10.1086/324565. PMC 1235548. PMID 11704930.
- ↑ Roux-Buisson, Nathalie; Cacheux, Marine; Fourest-Lieuvin, Anne; Fauconnier, Jeremy; Brocard, Julie; Denjoy, Isabelle; Durand, Philippe; Guicheney, Pascale; Kyndt, Florence; Leenhardt, Antoine; Le Marec, Hervé (2012-06-15). "Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human". Human Molecular Genetics. 21 (12): 2759–2767. doi:10.1093/hmg/dds104. ISSN 0964-6906. PMC 3363337. PMID 22422768.
- 1 2 Faggioni M, Kryshtal DO, Knollmann BC (August 2012). "Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia". Pediatric Cardiology. 33 (6): 959–67. doi:10.1007/s00246-012-0256-1. PMC 3393815. PMID 22421959.
- ↑ Liu N, Priori SG (January 2008). "Disruption of calcium homeostasis and arrhythmogenesis induced by mutations in the cardiac ryanodine receptor and calsequestrin". Cardiovascular Research. 77 (2): 293–301. doi:10.1093/cvr/cvm004. PMID 18006488.
- 1 2 Landstrom AP, Dobrev D, Wehrens XH (June 2017). "Calcium Signaling and Cardiac Arrhythmias". Circulation Research. 120 (12): 1969–1993. doi:10.1161/CIRCRESAHA.117.310083. PMC 5607780. PMID 28596175.
- 1 2 3 4 Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C (December 2013). "HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013". Heart Rhythm. 10 (12): 1932–63. doi:10.1016/j.hrthm.2013.05.014. PMID 24011539.
- ↑ Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, Skanes AC, Krahn AD (December 2011). "How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia". Circulation: Arrhythmia and Electrophysiology. 4 (6): 958–64. doi:10.1161/CIRCEP.111.965947. PMID 22203660.
- ↑ Semsarian C, Ingles J (October 2016). "Molecular autopsy in victims of inherited arrhythmias". Journal of Arrhythmia. 32 (5): 359–365. doi:10.1016/j.joa.2015.09.010. PMC 5063264. PMID 27761159.
- 1 2 3 4 5 6 7 Mazzanti, Andrea; Kukavica, Deni; Trancuccio, Alessandro; Memmi, Mirella; Bloise, Raffaella; Gambelli, Patrick; Marino, Maira; Ortíz-Genga, Martín; Morini, Massimo; Monteforte, Nicola; Giordano, Umberto (2022-03-30). "Outcomes of Patients With Catecholaminergic Polymorphic Ventricular Tachycardia Treated With β-Blockers". JAMA Cardiology. 7 (5): 504–512. doi:10.1001/jamacardio.2022.0219. ISSN 2380-6583. PMC 8968697. PMID 35353122.
- 1 2 Lieve KV, Wilde AA, van der Werf C (May 2016). "The Role of Flecainide in the Management of Catecholaminergic Polymorphic Ventricular Tachycardia". Arrhythmia & Electrophysiology Review. 5 (1): 45–9. doi:10.15420/aer.2016.3.3. PMC 4939313. PMID 27403293.
- ↑ Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, Belhassen B, Viskin S (September 2007). "Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia". Heart Rhythm. 4 (9): 1149–54. doi:10.1016/j.hrthm.2007.05.017. PMID 17765612.
- ↑ Shivkumar K, Ajijola OA, Anand I, Armour JA, Chen PS, Esler M, De Ferrari GM, Fishbein MC, Goldberger JJ, Harper RM, Joyner MJ, Khalsa SS, Kumar R, Lane R, Mahajan A, Po S, Schwartz PJ, Somers VK, Valderrabano M, Vaseghi M, Zipes DP (July 2016). "Clinical neurocardiology defining the value of neuroscience-based cardiovascular therapeutics". The Journal of Physiology. 594 (14): 3911–54. doi:10.1113/JP271870. PMC 4945719. PMID 27114333.
- 1 2 Schwartz PJ, De Ferrari GM, Pugliese L (June 2017). "Cardiac sympathetic denervation 100years later: Jonnesco would have never believed it". International Journal of Cardiology. 237: 25–28. doi:10.1016/j.ijcard.2017.03.020. PMID 28318666.
- ↑ Dhindwal, Sonali; Lobo, Joshua; Cabra, Vanessa; Santiago, Demetrio J.; Nayak, Ashok R.; Dryden, Kelly; Samsó, Montserrat (2017-05-23). "A cryo-EM–based model of phosphorylation- and FKBP12.6-mediated allosterism of the cardiac ryanodine receptor". Science Signaling. 10 (480): eaai8842. doi:10.1126/scisignal.aai8842. ISSN 1945-0877. PMID 28536302. S2CID 1931284.
- ↑ Swan H, Piippo K, Viitasalo M, Heikkilä P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L (December 1999). "Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts". Journal of the American College of Cardiology. 34 (7): 2035–42. doi:10.1016/S0735-1097(99)00461-1. PMID 10588221.
- ↑ Behere SP, Weindling SN (2016). "Catecholaminergic polymorphic ventricular tachycardia: An exciting new era". Annals of Pediatric Cardiology. 9 (2): 137–46. doi:10.4103/0974-2069.180645. PMC 4867798. PMID 27212848.