Classical nucleation theory (CNT) is the most common theoretical model used to quantitatively study the kinetics of nucleation.[1][2][3][4]
Nucleation is the first step in the spontaneous formation of a new thermodynamic phase or a new structure, starting from a state of metastability. The kinetics of formation of the new phase is frequently dominated by nucleation, such that the time to nucleate determines how long it will take for the new phase to appear. The time to nucleate can vary by orders of magnitude, from negligible to exceedingly large, far beyond reach of experimental timescales. One of the key achievements of classical nucleation theory is to explain and quantify this immense variation.[5]
Description
The central result of classical nucleation theory is a prediction for the rate of nucleation , in units of (number of events)/(volume·time). For instance, a rate in a supersaturated vapor would correspond to an average of 1000 droplets nucleating in a volume of 1 cubic meter in 1 second.


The CNT prediction for is[3]

where
- is the free energy cost of the nucleus at the top of the nucleation barrier, and is the average thermal energy with the absolute temperature and the Boltzmann constant.
- is the number of nucleation sites.
- is the rate at which molecules attach to the nucleus.
- is the Zeldovich factor, (named after Yakov Zeldovich[6]) which gives the probability that a nucleus at the top of the barrier will go on to form the new phase, rather than dissolve.







This expression for the rate can be thought of as a product of two factors: the first, , is the number of nucleation sites multiplied by the probability that a nucleus of critical size has grown around it. It can be interpreted as the average, instantaneous number of nuclei at the top of the nucleation barrier. Free energies and probabilities are closely related by definition.[7] The probability of a nucleus forming at a site is proportional to . So if is large and positive the probability of forming a nucleus is very low and nucleation will be slow. Then the average number will be much less than one, i.e., it is likely that at any given time none of the sites has a nucleus.

![\exp[-\Delta G^*/kT]](../I/2aa851977e60d3d4673586f9d4f6d00a716934a7.svg)
The second factor in the expression for the rate is the dynamic part, . Here, expresses the rate of incoming matter and is the probability that a nucleus of critical size (at the maximum of the energy barrier) will continue to grow and not dissolve. The Zeldovich factor is derived by assuming that the nuclei near the top of the barrier are effectively diffusing along the radial axis. By statistical fluctuations, a nucleus at the top of the barrier can grow diffusively into a larger nucleus that will grow into a new phase, or it can lose molecules and shrink back to nothing. The probability that a given nucleus goes forward is .

Taking into consideration kinetic theory and assuming that there is the same transition probability in each direction, it is known that . As determines the hopping rate, the previous formula can be rewritten in terms of the mean free path and the mean free time . Consequently, a relation of in terms of the diffusion coefficient is obtained


.

Further considerations can be made in order to study temperature dependence. Therefore, Einstein-Stokes relation is introduced under the consideration of a spherical shape
, where is the material's viscosity.


Considering the last two expressions, it is seen that . If , being the melting temperature, the ensemble gains high velocity and makes and to increase and hence, decreases. If , the ensemble has a low mobility, which makes to decrease as well.




To see how this works in practice we can look at an example. Sanz and coworkers[8] have used computer simulation to estimate all the quantities in the above equation, for the nucleation of ice in liquid water. They did this for a simple but approximate model of water called TIP4P/2005. At a supercooling of 19.5 °C, i.e., 19.5 °C below the freezing point of water in their model, they estimate a free energy barrier to nucleation of ice of . They also estimate a rate of addition of water molecules to an ice nucleus near the top of the barrier of and a Zeldovich factor . The number of water molecules in 1 m3 of water is approximately 1028. These leads to the prediction , which means that on average one would have to wait 1083s (1076 years) to see a single ice nucleus forming in 1 m3 of water at -20 °C!




This is a rate of homogeneous nucleation estimated for a model of water, not real water — in experiments one cannot grow nuclei of water and so cannot directly determine the values of the barrier , or the dynamic parameters such as , for real water. However, it may be that indeed the homogeneous nucleation of ice at temperatures near -20 °C and above is extremely slow and so that whenever water freezes at temperatures of -20 °C and above this is due to heterogeneous nucleation, i.e., the ice nucleates in contact with a surface.
Homogeneous nucleation
Homogeneous nucleation is much rarer than heterogeneous nucleation.[1][9] However, homogeneous nucleation is simpler and easier to understand than heterogeneous nucleation, so the easiest way to understand heterogeneous nucleation is to start with homogeneous nucleation. So we will outline the CNT calculation for the homogeneous nucleation barrier .


To understand if nucleation is fast or slow, , the Gibbs free energy change as a function of the size of the nucleus, needs to be calculated. The classical theory[10] assumes that even for a microscopic nucleus of the new phase, we can write the free energy of a droplet as the sum of a bulk term that is proportional to the volume of the nucleus, and a surface term, that is proportional to its surface area



The first term is the volume term, and as we are assuming that the nucleus is spherical, this is the volume of a sphere of radius . is the difference in free energy per unit of volume between the phase that nucleates and the thermodynamic phase nucleation is occurring in. For example, if water is nucleating in supersaturated air, then is the free energy per unit of volume of water minus that of supersaturated air at the same pressure. As nucleation only occurs when the air is supersaturated, is always negative. The second term comes from the interface at surface of the nucleus, which is why it is proportional to the surface area of a sphere. is the surface tension of the interface between the nucleus and its surroundings, which is always positive.



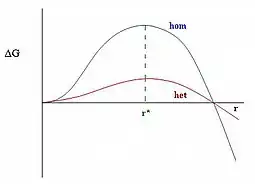
For small the second surface term dominates and . The free energy is the sum of an and terms. Now the terms varies more rapidly with than the term, so as small the term dominates and the free energy is positive while for large , the term dominates and the free energy is negative. This shown in the figure to the right. Thus at some intermediate value of , the free energy goes through a maximum, and so the probability of formation of a nucleus goes through a minimum. There is a least-probable nucleus size, i.e., the one with the highest value of



where
![{\displaystyle \left[{\frac {dG}{dr}}\right]_{r=r_{c}}=0\implies r_{c}={\frac {2\sigma }{|\Delta g_{v}|}}}](../I/0b24135d9b873e1ff6e4f63bbc7f373441d7fa52.svg)
Addition of new molecules to nuclei larger than this critical radius, , decreases the free energy, so these nuclei are more probable. The rate at which nucleation occurs is then limited by, i.e., determined by the probability, of forming the critical nucleus. This is just the exponential of minus the free energy of the critical nucleus , which is


This is the free energy barrier needed in the CNT expression for above.
In the discussion above, we assumed the growing nucleus to be three-dimensional and spherical. Similar equations can be set up for other dimensions and/or other shapes, using the appropriate expressions for the analogues of volume and surface area of the nucleus. One will then find out that any non-spherical nucleus has a higher barrier height than the corresponding spherical nucleus. This can be understood from the fact that a sphere has the lowest possible surface area to volume ratio, thereby minimizing the (unfavourable) surface contribution with respect to the (favourable) bulk volume contribution to the free energy. Assuming equal kinetic prefactors, the fact that is higher for non-spherical nuclei implies that their formation rate is lower. This explains why in homogeneous nucleation usually only spherical nuclei are taken into account.
From an experimental standpoint, this theory grants tuning of the critical radius through the dependence of on temperature. The variable , described above, can be expressed as

where is the melting point and is the enthalpy of formation for the material. Furthermore, the critical radius can be expressed as


revealing a dependence of reaction temperature. Thus as you increase the temperature near , the critical radius will increase. Same happens when you move away from the melting point, the critical radius and the free energy decrease.
Heterogeneous nucleation
.svg.png.webp)
Unlike homogeneous nucleation, heterogeneous nucleation occurs on a surface or impurity. It is much more common than homogeneous nucleation. This is because the nucleation barrier for heterogeneous nucleation is much lower than for homogeneous nucleation. To see this, note that the nucleation barrier is determined by the positive term in the free energy , which is proportional to the total exposed surface area of a nucleus. For homogeneous nucleation the surface area is simply that of a sphere. For heterogeneous nucleation, however, the surface area is smaller since part of the nucleus boundary is accommodated by the surface or impurity onto which it is nucleating.[11]
There are several factors which determine the precise reduction in the exposed surface area.[11] As shown in a diagram on the left, these factors include the size of the droplet, the contact angle, , between the droplet and surface, and the interactions at the three phase interfaces: liquid-solid, solid-vapor, and liquid-vapor.

The free energy needed for heterogeneous nucleation, , is equal to the product of homogeneous nucleation, , and a function of the contact angle, :



.

The schematic to the right illustrates the decrease in the exposed surface area of the droplet as the contact angle decreases. Deviations from a flat interface decrease the exposed surface even further: there exist expressions for this reduction for simple surface geometries.[12] In practice, this means that nucleation will tend to occur on surface imperfections.
Statistical mechanical treatment
The classical nucleation theory hypothesis for the form of can be examined more rigorously using the tools of statistical mechanics.[13] Specifically, the system is modeled as a gas of non-interacting clusters in the grand canonical ensemble. A state of metastable equilibrium is assumed, such that the methods of statistical mechanics hold at least approximately.[14] The grand partition function is[15]

Here the inner summation is over all microstates which contain exactly particles. It can be decomposed into contributions from each possible combination of clusters which results in total particles.[16] For instance,



where is the configuration integral of a cluster with particles and potential energy :




The quantity is the thermal de Broglie wavelength of the particle, which enters due to the integration over the momentum degrees of freedom. The inverse factorials are included to compensate for overcounting, since particles and clusters alike are assumed indistinguishable.


More compactly,
- .
![{\displaystyle Q=\exp \left[\sum _{n=1}^{\infty }q_{n}z^{n}\right]}](../I/f7814c135b28dd71e6778c830e2f4d3272dfaab3.svg)
Then, by expanding in powers of , the probability of finding exactly clusters which each has particles is






The number density of -clusters can therefore be calculated as


This is also called the cluster size distribution.
The grand potential is equal to , which, using the thermodynamic relationship , leads to the following expansion for the pressure:




If one defines the right hand side of the above equation as the function , then various other thermodynamic quantities can be calculated in terms of derivatives of with respect to .[17]



The connection with the simple version of the theory is made by assuming perfectly spherical clusters, in which case depends only on , in the form

where is the binding energy of a single particle in the interior of a cluster, and is the excess energy per unit area of the cluster surface. Then, , and the cluster size distribution is



![{\displaystyle \rho _{n}\propto e^{-\beta [-(E_{0}+\mu )n+wn^{1/2}]},}](../I/a642d75d1202f83e33ec1e37988b3cd3a700dfde.svg)
which implies an effective free energy landscape , in agreement with the form proposed by the simple theory.

On the other hand, this derivation reveals the significant approximation in assuming spherical clusters with . In reality, the configuration integral contains contributions from the full set of particle coordinates , thus including deviations from spherical shape as well as cluster degrees of freedom such as translation, vibration, and rotation. Various attempts have been made to include these effects in the calculation of , although the interpretation and application of these extended theories has been debated.[5][18][19] A common feature is the addition of a logarithmic correction to , which plays an important role near the critical point of the fluid.[20]




Limitations
Classical nucleation theory makes a number of assumptions which limit its applicability. Most fundamentally, in the so-called capillarity approximation it treats the nucleus interior as a bulk, incompressible fluid and ascribes to the nucleus surface the macroscopic interfacial tension , even though it is not obvious that such macroscopic equilibrium properties apply to a typical nucleus of, say, 50 molecules across.[21][22] In fact, it has been shown that the effective surface tension of small droplets is smaller than that of the bulk liquid.[23]
In addition, the classical theory places restrictions on the kinetic pathways by which nucleation occurs, assuming clusters grow or shrink only by single particle adsorption/emission. In reality, merging and fragmentation of entire clusters cannot be excluded as important kinetic pathways in some systems. Particularly in dense systems or near the critical point – where clusters acquire an extended and ramified structure – such kinetic pathways are expected to contribute significantly.[23] The behavior near the critical point also suggests the inadequacy, at least in some cases, of treating clusters as purely spherical.[24]
Various attempts have been made to remedy these limitations and others by explicitly accounting for the microscopic properties of clusters. However, the validity of such extended models is debated. One difficulty is the exquisite sensitivity of the nucleation rate to the free energy : even small discrepancies in the microscopic parameters can lead to enormous changes in the predicted nucleation rate. This fact makes first-principles predictions nearly impossible. Instead, models must be fit directly to experimental data, which limits the ability to test their fundamental validity.[25]
Comparison with simulation and experiment
For simple model systems, modern computers are powerful enough to calculate exact nucleation rates numerically. An example is the nucleation of the crystal phase in a system of hard spheres, which is a simple model of colloids consisting of perfectly hard spheres in thermal motion. The agreement of CNT with the simulated rates for this system confirms that the classical theory is a reasonable approximation.[26] For simple models CNT works quite well; however it is unclear if it describes complex (e.g., molecular) systems equally well. For example, in the context of vapor to liquid nucleation, the CNT predictions for the nucleation rate are incorrect by several orders of magnitude on an absolute scale — that is, without renormalizing with respect to experimental data. Nevertheless, certain variations on the classical theory have been claimed to represent the temperature dependence adequately, even if the absolute magnitude is inaccurate.[27] Jones et al. computationally explored the nucleation of small water clusters using a classical water model. It was found that CNT could describe the nucleation of clusters of 8-50 water molecules well, but failed to describe smaller clusters.[28] Corrections to CNT, obtained from higher accuracy methods such as quantum chemical calculations, may improve the agreement with experiment.[29]
References
- 1 2 H. R. Pruppacher and J. D. Klett, Microphysics of Clouds and Precipitation, Kluwer (1997)
- ↑ P.G. Debenedetti, Metastable Liquids: Concepts and Principles, Princeton University Press (1997)
- 1 2 Sear, R. P. (2007). "Nucleation: theory and applications to protein solutions and colloidal suspensions". J. Phys.: Condens. Matter. 19 (3): 033101. Bibcode:2007JPCM...19c3101S. CiteSeerX 10.1.1.605.2550. doi:10.1088/0953-8984/19/3/033101. S2CID 4992555.
- ↑ Kreer, Markus (1993). "Classical Becker‐Döring cluster equations: Rigorous results on metastability and long‐time behaviour". Annalen der Physik. 505 (4): 398–417. Bibcode:1993AnP...505..398K. doi:10.1002/andp.19935050408.
- 1 2 Oxtoby, David W. (1992), "Homogeneous nucleation: theory and experiment", Journal of Physics: Condensed Matter, 4 (38): 7627–7650, Bibcode:1992JPCM....4.7627O, doi:10.1088/0953-8984/4/38/001, S2CID 250827558
- ↑ Zeldovich, Y. B. (1943). On the theory of new phase formation: cavitation. Acta Physicochem., USSR, 18, 1.
- ↑ Frenkel, Daan; Smit, Berent (2001). Understanding Molecular Simulation, Second Edition: From Algorithms to Applications. p. Academic Press. ISBN 978-0122673511.
- ↑ Sanz, Eduardo; Vega, Carlos; Espinosa, J. R.; Cabellero-Bernal, R.; Abascal, J. L. F.; Valeriani, Chantal (2013). "Homogeneous Ice Nucleation at Moderate Supercooling from Molecular Simulation". Journal of the American Chemical Society. 135 (40): 15008–15017. arXiv:1312.0822. Bibcode:2013arXiv1312.0822S. doi:10.1021/ja4028814. PMID 24010583. S2CID 2304292.
- ↑ Sear, Richard P. (2014). "Quantitative Studies of Crystal Nucleation at Constant Supersaturation: Experimental Data and Models". CrystEngComm. 16 (29): 6506–6522. doi:10.1039/C4CE00344F.
- ↑ F. F. Abraham (1974) Homogeneous nucleation theory (Academic Press, NY).
- 1 2 Liu, X. Y. (31 May 2000). "Heterogeneous nucleation or homogeneous nucleation?". The Journal of Chemical Physics. 112 (22): 9949–9955. Bibcode:2000JChPh.112.9949L. doi:10.1063/1.481644. ISSN 0021-9606.
- ↑ Sholl, C. A.; N. H. Fletcher (1970). "Decoration criteria for surface steps". Acta Metall. 18 (10): 1083–1086. doi:10.1016/0001-6160(70)90006-4. hdl:1885/213299.
- ↑ The discussion which follows draws from Kalikmanov (2001), unless noted otherwise.
- ↑ Kalikmanov, V.I. (2013), Nucleation Theory, Lecture Notes in Physics, vol. 860, Springer Netherlands, pp. 17–19, Bibcode:2013LNP...860.....K, doi:10.1007/978-90-481-3643-8, ISBN 978-90-481-3643-8, ISSN 0075-8450
- ↑ Kardar, Mehran (2007), Statistical Physics of Particles, Cambridge University Press, p. 118, ISBN 978-0-521-87342-0
- ↑ Kalikmanov, V.I. (2001), Statistical Physics of Fluids: Basic Concepts and Applications, Springer-Verlag, pp. 170–172, ISBN 978-3-540-417-47-7, ISSN 0172-5998
- ↑ Kalikmanov (2001) pp. 172-173
- ↑ Kiang, C. S. and Stauffer, D. and Walker, G. H. and Puri, O. P. and Wise, J. D. and Patterson, E. M., C.S.; Stauffer, D.; Walker, G. H.; Puri, O.P.; Wise, J.D.; Patterson, E.M. (1971), "A Reexamination of Homogeneous Nucleation Theory", Journal of the Atmospheric Sciences, 28 (7): 1222–1232, Bibcode:1971JAtS...28.1222K, doi:10.1175/1520-0469(1971)028<1222:AROHNT>2.0.CO;2
{{citation}}: CS1 maint: multiple names: authors list (link) - ↑ Reguera, D.; Rubí, J.M. (2001), "Nonequilibrium translational-rotational effects in nucleation", The Journal of Chemical Physics, 115 (15): 7100–7106, arXiv:cond-mat/0109270, Bibcode:2001JChPh.115.7100R, doi:10.1063/1.1405122, S2CID 95887792
- ↑ Sator, N. (2003), "Clusters in simple fluids", Physics Reports, 376 (1): 1–39, arXiv:cond-mat/0210566, Bibcode:2003PhR...376....1S, doi:10.1016/S0370-1573(02)00583-5, ISSN 0370-1573, S2CID 119492597
- ↑ Kalikmanov (2013), p. 21
- ↑ Oxtoby (1992), p. 7631
- 1 2 Kiang, et al (1971)
- ↑ Sator (2003)
- ↑ Oxtoby (1992), p. 7638–7640
- ↑ Auer, S.; D. Frenkel (2004). "Numerical prediction of absolute crystallization rates in hard-sphere colloids" (PDF). J. Chem. Phys. 120 (6): 3015–29. Bibcode:2004JChPh.120.3015A. doi:10.1063/1.1638740. hdl:1874/12074. PMID 15268449. S2CID 15747794.
- ↑ Fladerer, Alexander; Strey, Reinhard (2006-04-28). "Homogeneous nucleation and droplet growth in supersaturated argon vapor: The cryogenic nucleation pulse chamber". The Journal of Chemical Physics. AIP Publishing. 124 (16): 164710. Bibcode:2006JChPh.124p4710F. doi:10.1063/1.2186327. ISSN 0021-9606. PMID 16674160.
- ↑ Merikanto, Joonas; Zapadinsky, Evgeni; Lauri, Antti; Vehkamäki, Hanna (4 April 2007). "Origin of the Failure of Classical Nucleation Theory: Incorrect Description of the Smallest Clusters". Physical Review Letters. 98 (14): 145702. Bibcode:2007PhRvL..98n5702M. doi:10.1103/PhysRevLett.98.145702. PMID 17501289.
- ↑ Temelso, Berhane; Morrell, Thomas E.; Shields, Robert M.; Allodi, Marco A.; Wood, Elena K.; Kirschner, Karl N.; Castonguay, Thomas C.; Archer, Kaye A.; Shields, George C. (22 February 2012). "Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications". The Journal of Physical Chemistry A. 116 (9): 2209–2224. Bibcode:2012JPCA..116.2209T. doi:10.1021/jp2119026. PMID 22296037.