DNA end resection, also called 5′–3′ degradation, is a biochemical process where the blunt end of a section of double-stranded DNA (dsDNA) is modified by cutting away some nucleotides from the 5' end to produce a 3' single-stranded sequence.[1][2] The presence of a section of single-stranded DNA (ssDNA) allows the broken end of the DNA to line up accurately with a matching sequence, so that it can be accurately repaired.[1]
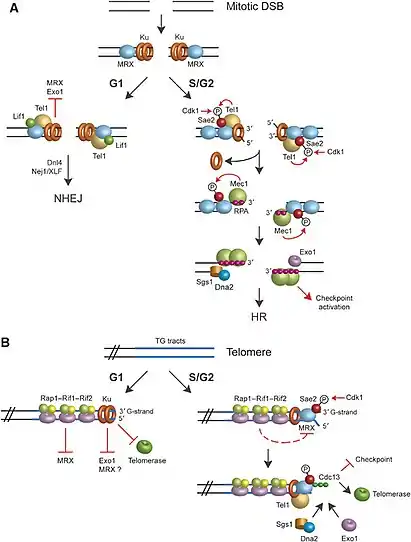
Double-strand breaks (DSBs) can occur at any phase of the cell cycle causing DNA end resection and repair activities to take place, but they are also normal intermediates in mitosis recombination.[3] Furthermore, the natural ends of the linear chromosomes resemble DSBs, and although DNA breaks can cause damage to the integrity of genomic DNA, the natural ends are packed into complex specialized DNA protective packages called telomeres that prevent DNA repair activities.[3][4] Telomeres and mitotic DSBs have different functionality, but both experience the same 5′–3′ degradation process.
Background
A double-strand break is a kind of DNA damage in which both strands in the double helix are severed. DSBs only occur during DNA replication of the cell cycle. Furthermore, DSBs can lead to genome rearrangements and instability.[3] Cases where two complementary strands are linked at the point of the DSB have potential to be catastrophic, such that the cell will not be able to complete mitosis when it next divides, and will either die or, in rare cases, undergo chromosomal loss, duplications, and even mutations.[5][6] Three mechanisms exist to repair DSBs: non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination HR.[7][8][9] Of these, only NHEJ does not rely on DNA end resection.[2]
Mechanism
Accurate repair of DSBs are essential in the upkeep of genome integrity. From the three mechanisms that exists to repair DSBs, NHEJ and HR repair mechanisms are the dominant pathways.[4] Several highly conservative proteins trigger the DNA Damage Checkpoint for detection of DSBs ensuing repair by either NHEJ or HR repair pathways.[3] NHEJ mechanism functions in ligating two different DSBs with high fidelity, while HR relies on a homologous template to repair DSB ends.[3][4]
DNA end resection in the HR pathway only occurs at two specific phases: S and G2 phases.[4][3] Since HR pathway requires sister chromatids for activation, this event only happens in the G2 and S phases of the cell cycle during replication.[3][4] DSBs that have not begun DNA end resection can be ligated by NHEJ pathway, but resection of a few nucleotides inhibits the NHEJ pathway and commits' DNA repair by the HR pathway.[10] The NHEJ pathway is involved throughout the cell cycle, but it is critical to DNA repair during the G1 phase.[3][10] In G1 phase there is no sister chromatids to repair DSBs via the HR pathway making the NHEJ pathway a critical repair mechanism.[3][10]
Before resection can take place, the break needs to be detected. In animals, this detection is done by PARP1;[11] similar systems exist in other eukaryotes: in plants, PARP2 seems to play this role.[12] PARP binding then recruits the MRN complex to the breakage site.[13] This is a highly conserved complex consisting of Mre11, Rad50 and NBS1 (known as Nibrin[14] in mammals, or Xrs2 in yeast, where this complex is called the MRX complex).
Before resection can start, CtBP1-interacting protein (CtIP) needs to bind to the MRN complex so that the first phase of resection can begin, namely short-range end resection.[7][15][16] After phosphorylated CtIP binds, the Mre11 subunit is able to cut the 5'-terminated strand endonucleolytically, probably about 300 base pairs from the end,[15][16] and then acts as a 3'→5' exonuclease to strip away the end of the 5' strand.[16]
Resection of telomere DSBs
Linear chromosomes are packed into complex specialized DNA protective packages called telomeres.[3][4] The structure of telomeres is highly conserve and is organized in multiple short tandem DNA repeats.[3] Telomeres and DSBs have different functionality, such that telomeres prevent DNA repair activities.[3] During telomeric DNA replication in the S/G2 and G1 phases of the cell cycle, the 3' lagging strand leaves a short overhang called a G-tail.[4][3] Telomeric DNA ends at the 3' G tail end because the 3' lagging strand extends without its complementary 5' C leading strand. The G tail provide a major function to telomeric DNA such that the G tails control telomere homeostasis.[4]
Telomeres in G1 phase
In the G1 phase of the cell cycle, the telomere-associated proteins RIF1, RIF2, and RAP2 bind to telomeric DNA and prevent access to the MRX complex.[4][3] Such process in S. Cerevisiae for example is negatively regulated by this activity. The MRX complex and the Ku complex bind simultaneously and independently to DSBs ends.[3][16] In the presence of the telomere-associated proteins, MRX fails to bind to the DSB ends while the Ku complex binds to DSB ends.[3] The bound Ku complex to the DSB ends protect the telomeres from nucleolytic degradation by exo1.[3] This results in an inhibition of telomerase elongation at the DSB ends and prevents further telomere action at the G1 phase of the cell cycle.[3][4]
Telomeres in the late S/G2 phase
In the late S/G2 phase of the cell cycle, the telomere-associated proteins RIF1, RIF2, and RAP2 exhibit their inhibitory effect by binding to telomeric DNA.[3] In the Late S/G2 phase, the protein kinase CDK1 (cyclin-dependent) promotes telomeric resection.[4][3] This control is exerted by cyclin-dependent kinases, which phosphorylate parts of the resection machinery.[15] This process alleviates the inhibitory effect of the telomere-associated proteins, and allows Cdc13 (a binding protein on both the lagging strand, and leading strand) to cover telomeric DNA.[15] The binding of cdc13 to DNA suppresses DNA damage checkpoint and allows resection to occur while allowing for telomerase elongation at the DSB ends.[3]
Resection of mitotic DSBs
One of the important regulatory controls in mitotic cells is deciding which specific DSB repair pathway to take. Once a DSB is detected, the highly conserved complexes are recruited by the DNA ends.[2] If the cell is in the G1 phase of the cell cycle, the complex Ku prevents resection to occur and triggers the NHEJ pathway factors.[3] DSBs in the NHEJ pathway are ligated, a step in the NHEJ pathway that requires DNA ligase activity of Dnl4-Lif1/XRCC4 heterodimer and the Nej1/XLF protein.[3] This process results in error-prone religation of DSB ends at the G1 phase of the cell cycle.[3]
If the cells are in S/G2 phase, mitotic DSBs are controlled through Cdk1 activity and involves phosphorylation of Sae2 Ser267.[4][3] After phosphorylation occurs by Cdk1, MRX complex binds to dsDNA ends and generates short ssDNA that stretches in the 5' direction.[4][3] The 5' ssDNA continues resection by the activity of the helicase enzyme, Sgs1 enzyme, and the nucleases Exo1 and Dna2.[17] Involvement of Sae2 Sar267 in DSB processing is highly conserved throughout eukaryotes, such that the Sae2 along with the MRX complex are involved in two major functions: single-strand annealing, and processing of hairpin DNA structures.[4] Like all ssDNA in the nucleus, the resected region is first coated by Replication protein A (RPA) complex,[18][15] but RPA is then replaced with RAD51 to form a nucleoprotein filament which can take part in the search for a matching region, allowing HR to take place.[15] The 3' ssDNA coated by a RPA promotes the recruitment of Mec1. Mec1 further phosphorylates Sae2 along with cdk1. The resulting phosphorylation by Sae2 by Mec1 helps increase the effect of resection and this in turn leads to the DNA damage checkpoint activation.[4][3]
Regulators
The pathway of choice in DNA repair is highly regulated to guarantee that cells in the S/G2 and G1 phase use the appropriate mechanism. Regulators in both the NHEJ and HR pathway mediate the appropriate DNA repair response pathway.[3] Furthermore, recent studies into DNA repair show that regulation of DNA end resection is governed by the activity of cdk1 in the cell replication cycle.[3][19]
NHEJ pathway
DNA end resection is key in determining the correct pathway in NHEJ. For NHEJ pathway to occur, positive regulators such as the Ku and MRX complex mediate recruitment of other NHEJ-associated proteins such as Tel1, Lif1, Dnl4, and Nej1.[3] Since NHEJ does not rely on end resection, NHEJ could only happen in the G1 phase of the cell cycle.[3][10] Both Ku and NHEJ-associated proteins prevent initiation of resection.
Resection ensures that DSBs are not repaired by NHEJ (which joins broken DNA ends together without ensuring that they match), but rather by methods based on homology (matching DNA sequences). Cyclin-dependent protein kinase such as cdk1 in yeast serves as a negative regulator of the NHEJ pathway.[3] Any activity associated with the presence of cyclin dependent protein kinases inhibit the NHEJ pathway[3]
Positive regulators
The presence of a ssDNA allows the broken end of the DNA to line up accurately with a matching sequence, so that it can be accurately repaired.[1] For HR pathway to occur in the S and G2 phases of the cell cycle, availability of a sister chromatid is required.[3][4] 5′–3′ resection automatically links a DSB to the HR pathway.[2] Cyclin-dependent protein kinase such as cdk1 serve as a positive regulator of the HR pathway.[4][3] This positive regulator promotes 5′–3′ nucleolytic degradation of DNA ends. Along with cdk1, the MRX complex, B1 cyclin, and Spo11-induced DSBs serve as a positive regulators to the HR pathway.[17][19]
See also
References
- 1 2 3 Jimeno S, Mejías-Navarro F, Prados-Carvajal R, Huertas P (2019). "Controlling the balance between chromosome break repair pathways". Advances in Protein Chemistry and Structural Biology. Elsevier. 115: 95–134. doi:10.1016/bs.apcsb.2018.10.004. ISBN 9780128155592. PMID 30798939. S2CID 73459973.
- 1 2 3 4 Liu T, Huang J (June 2016). "DNA End Resection: Facts and Mechanisms". Genomics, Proteomics & Bioinformatics. 14 (3): 126–130. doi:10.1016/j.gpb.2016.05.002. PMC 4936662. PMID 27240470.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 Longhese MP, Bonetti D, Manfrini N, Clerici M (September 2010). "Mechanisms and regulation of DNA end resection". The EMBO Journal. 29 (17): 2864–2874. doi:10.1038/emboj.2010.165. PMC 2944052. PMID 20647996.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Mimitou EP, Symington LS (September 2009). "DNA end resection: many nucleases make light work". DNA Repair. 8 (9): 983–995. doi:10.1016/j.dnarep.2009.04.017. PMC 2760233. PMID 19473888.
- ↑ Bjorksten J, Acharya PV, Ashman S, Wetlaufer DB (July 1971). "Gerogenic fractions in the tritiated rat". Journal of the American Geriatrics Society. 19 (7): 561–574. doi:10.1111/j.1532-5415.1971.tb02577.x. PMID 5106728. S2CID 33154242.
- ↑ Acharya PV (1972). "The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides". Johns Hopkins Medical Journal. Supplement (1): 254–260. PMID 5055816.
- 1 2 Huertas P (January 2010). "DNA resection in eukaryotes: deciding how to fix the break". Nature Structural & Molecular Biology. 17 (1): 11–16. doi:10.1038/nsmb.1710. PMC 2850169. PMID 20051983.
- ↑ Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA (September 2005). "Modulation of DNA end joining by nuclear proteins". The Journal of Biological Chemistry. 280 (36): 31442–31449. doi:10.1074/jbc.M503776200. PMID 16012167.
- ↑ Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R, eds. (2004). Molecular Biology of the Gene (5th ed.). Pearson Benjamin Cummings; CSHL Press. Ch. 9, 10. OCLC 936762772.
- 1 2 3 4 Daley JM, Laan RL, Suresh A, Wilson TE (August 2005). "DNA joint dependence of pol X family polymerase action in nonhomologous end joining". The Journal of Biological Chemistry. 280 (32): 29030–29037. doi:10.1074/jbc.M505277200. PMID 15964833.
- ↑ Ray Chaudhuri A, Nussenzweig A (October 2017). "The multifaceted roles of PARP1 in DNA repair and chromatin remodelling". Nature Reviews. Molecular Cell Biology. 18 (10): 610–621. doi:10.1038/nrm.2017.53. PMC 6591728. PMID 28676700.
- ↑ Song J, Keppler BD, Wise RR, Bent AF (May 2015). "PARP2 Is the Predominant Poly(ADP-Ribose) Polymerase in Arabidopsis DNA Damage and Immune Responses". PLOS Genetics. 11 (5): e1005200. doi:10.1371/journal.pgen.1005200. PMC 4423837. PMID 25950582.
- ↑ Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (January 2008). "PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites". The Journal of Biological Chemistry. 283 (2): 1197–1208. doi:10.1074/jbc.M706734200. PMID 18025084.
- ↑ Uhrhammer N, Bay JO, Gatti RA (October 2002). "NBN (Nijmegen breakage syndrome 1)". Atlas of Genetics and Cytogenetics in Oncology and Haematology - NBS1. Archived from the original on 2006-09-29. Retrieved 2008-02-12.
- 1 2 3 4 5 6 Casari E, Rinaldi C, Marsella A, Gnugnoli M, Colombo CV, Bonetti D, Longhese MP (2019). "Processing of DNA Double-Strand Breaks by the MRX Complex in a Chromatin Context". Frontiers in Molecular Biosciences. 6: 43. doi:10.3389/fmolb.2019.00043. PMC 6567933. PMID 31231660.
- 1 2 3 4 Pinto C, Anand R, Cejka P (2018). "Methods to Study DNA End Resection II: Biochemical Reconstitution Assays". Mechanisms of DNA Recombination and Genome Rearrangements: Methods to Study Homologous Recombination. Methods in Enzymology. Vol. 600. pp. 67–106. doi:10.1016/bs.mie.2017.11.009. ISBN 9780128144299. PMID 29458776.
- 1 2 Xue C, Wang W, Crickard JB, Moevus CJ, Kwon Y, Sung P, Greene EC (March 2019). "Regulatory control of Sgs1 and Dna2 during eukaryotic DNA end resection". Proceedings of the National Academy of Sciences of the United States of America. 116 (13): 6091–6100. Bibcode:2019PNAS..116.6091X. doi:10.1073/pnas.1819276116. PMC 6442620. PMID 30850524.
- ↑ Chen C, ed. (2013). New research directions in DNA repair. Croatia: InTech. ISBN 978-953-51-1114-6. OCLC 957280914.
- 1 2 Poon RY (2016-01-01). "Mitotic Catastrophe". In Bradshaw RA, Stahl PD (eds.). Encyclopedia of Cell Biology. Waltham: Academic Press. pp. 399–403. ISBN 978-0-12-394796-3.