Epigenetic regulation of neurogenesis is the role that epigenetics (hertitable characteristics that do not involve changes in DNA sequence) plays in the regulation of neurogenesis (the production of neurons from neural stem cells).
Epigenetics is the study of heritable changes in gene expression which do not result from modifications to the sequence of DNA. Neurogenesis is the mechanism for neuron proliferation and differentiation. It entails many different complex processes which are all time and order dependent.[1]
Processes such as neuron proliferation, fate specification, differentiation, maturation, and functional integration of newborn cells into existing neuronal networks are all interconnected.[2] In the past decade many epigenetic regulatory mechanisms have been shown to play a large role in the timing and determination of neural stem cell lineages.[1]
Mechanisms
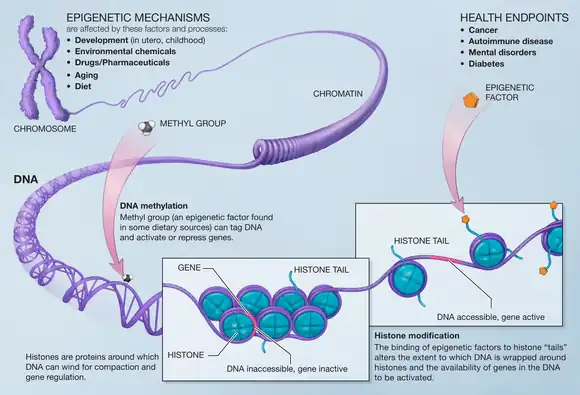
Three important methods of epigenetic regulation include histone modification, DNA methylation and demethylation, and microRNA (miRNA) expression. Histones keep the DNA of the eukaryotic cell tightly packaged through charge interactions between the positive charge on the histone tail and the negative charge of the DNA, as well as between histone tails of nearby nucleosomes. While there are many different types of histone modifications, in neural epigenetics there are two primary mechanisms which have been explored: histone methylation and histone acetylation.[1][3] In the former, methyl groups are either added or removed to the histone altering its structure and exposing chromatin and leading to gene activation or deactivation. In the latter, histone acetylation causes the histone to hold the DNA more loosely, allowing for more gene activation. DNA methylation, in which methyl groups are added to cytosine or adenosine residues on the DNA, is a more lasting method of gene inactivation than histone modification, though is still reversible in some cases.[1][3] MicroRNAs are a small form of non-coding RNA (ncRNA) which often act as "fine-tuning" mechanisms for gene expression by repressing or inducing messenger RNA (mRNA) in neural cells but can also act directly with transcription factors to guide neurogenesis.[1][3][4][5]
Embryonic neurogenesis
Histone modifications
Neural stem cells are involved in the development of the cortex in a precise "inside out" manner with carefully controlled timing mechanisms. Early born neurons form deep layers in the cortex while newer born neurons form the upper layers. This timing program is seen in vitro as well as in vivo.[1][3] Mutant analysis has shown that histone methylation modulates the production of deep layer and upper layer neurons through epigenetic regulation. Specifically, deletion of a portion of the PRC2 complex, Ezh2, encoding histone methyltransferase, led to a twofold reduction of POU3F2/BRN2-expressing and SATB2-expressing upper layer neurons without affecting the number of neurons in layers V and VI . In mouse embryonic stem cell-derived neural progenitors, increased histone acetylation induced by the histone deacetylase (HDAC) inhibitor valproic acid not only induced neuronal differentiation, but also selectively enriched the upper layer neuronal population. Therefore, it has been proposed that HDAC inhibition promotes the progression of neuronal differentiation, leading to a fate-switch from deep-layer producing progenitors into upper-layer progenitors. However, the reasons behind this selective differentiation and timing control as a result of HDAC inhibition are not yet fully understood.[3]
DNA methylation
DNA methylation's critical nature to corticogenesis has been shown through knockout experiments in mice. When DNMT3b and DNMT1 were ablated separately in mouse embryos they died due to impairment of neural tube development. DNMT3a silencing did not cause embryonic lethality, but did result in a severe detriment in postnatal neurogenesis.[1][3] This is largely due to the timing in which these epigenetic mechanisms are active. DNMT3b is expressed in early neural progenitor cells and decrease as neural development proceeds and DNMT3a is barely detectable up until embryonic day 10 (E.10). However, at E.10, DNMT3a expression increases significantly from E13.5 and well into adulthood. In the postnatal forebrain, DNMT3a is expressed in the subventricular zone (SVZ) and the hippocampal dentate gyrus, the primary locations for adult neurogenesis.[1][2] The loss of DNMT3a in post natal neural progenitor cells leads to the down-regulation of neuronic genes such as Dlx2, Neurog2, and Sp8; but upregulation of genes involved in astroglial and oligodendroglial differentiation, indicating a role in the cell-fate switch from neurogenesis to gliogenesis. DNA demethylation, as well as methylation, of certain genes allows for neurogenesis to proceed in a time dependent manner. One such gene is Hes5, hypermethylated in E7.5 Embryos but completely demethylated by E9.5, which is one of the target genes in the Notch Signaling pathway. GCM1 and GCM2 demethylate the Hes5 promoter, allowing it to respond to NOTCH signaling and initiating the generation of neural stem cells.[1] Another example is the Gfap gene which is required for astrocyte differentiation. The ability to differentiate into glial cells is repressed in neural stem cells with a neuronal cell fate. This repression is due largely to an irresponsiveness of neural stem cells towards astrocyte-inducing stimulations. The neural stem cells are non-responsive due to hypermethylated DNA in the promoter regions of astrocyte genes such as Gfap. The STAT3 binding site in the promoter region of Gfap is hypermethylated at E11.5 and barely so at E14.5, at which point it is able to receive astrocyte inducing stimulations and begin cytokine-inducible astrocyte differentiation.[3]
miRNAs

Studies done by De Pietri Tonelli and Kawase-Koga have shown conditional knockout of Dicer, an enzyme largely used for miRNA synthesis, in mouse neocortex resulted in reduced cortical size, increased neuronal apoptosis, and deficient cortisol layering. Neuroepithelial cells and neuroprogenitor cells were not affected until E.14, at which point they also underwent apoptosis. This doesn't show which miRNAs were responsible for the varying factors affected, but it does show that there is a stage-specific requirement for miRNA expression in cortical development.[1][5][6][7] miR-124, the most abundant microRNA in the central nervous system, controls the lineage progression of subventricular zone neural progenitor cells into neuroblasts by suppressing protein production by targeting Sox9. Another major microRNA player is miR-9/9*. In embryonic neurogenesis miR-9 has been shown to regulate neuronal differentiation and self-renewal.[1][4][5] Ectopic expression of miR-9 in the developing mouse cortex led to premature neuronal differentiation and disrupted the migration of new neurons through targeting Foxg1.[1]
Contrary to the idea that microRNAs are only fine-tuning mechanisms, recent studies have shown that miR-9 and miR-124 can act together to guide fibroblasts into neural cells. Transcription factors and regulatory genes, such as Neurod1, Ascr1, and Myt1l, which were previously thought to be responsible for this phenomenon did not transform human fibroblasts in the absence of miR-9 and miR-124, but in the presence of the microRNAs and the absence of the transcription factors human fibroblast transformation proceeded, albeit in a less efficient manner.[1][4][5]
Adult neurogenesis
DNA methylation
Neurogenesis continues after development well through adulthood.[2] Growth arrest and DNA-damage-inducible, beta (GADD45b) is required for the demethylation of promoters of critical genes responsible for new-born neuron development such as brain-derived neurotrophic factor (BDNF) and basic fibroblast growth factor (FGF2).[8] As such, upregulation of GADD45b leads to increased demethylation, increased BDNF and FGF2, and ultimately more neural progenitor cells.[1][8]
Histone modifications via acetylation
Histone acetylation, deacetylation, as well as the inhibition of histone deacetylation mechanisms also play large roles in the proliferation and self-renewal of post-natal neural stem cells. DNA, which encodes genes within the genome, including those involved in adult neurogenesis, is packaged into chromatin. Chromatin itself is made up of nucleosome subunits, each consisting of two copies each of histone proteins H2A, H2B, H3 and H4. One of the primary roles acetylation plays in the regulation of gene expression is through the inhibition of adjacent nucleosome interactions. When the H4 histones are not acetylated, they are basic in nature, and insert into the acidic pocket of the H2A-H2B protein dimers in adjacent nucleosomes, leading to tight association between nucleosomes and further packing of the chromatin. Thus, acetylation causes the H4 histone to lose its basicity, and prevents nucleosome cross-linking.[9] This acetylation of histone tails additionally increase the affinity of chromatin remodeling enzyme complexes such as SWI–SNF and ISWI, which utilize ATP to produce nucleosome-free regions at promoter and enhancer sites.[10] This allows for greater ability of recognition of these sites by transcription factors, particularly TFIID which is the major transcription factor involved in transcription initiation. Furthermore, the acetylation of lysine residues on histone tails can be recognized by the TAF1 component of TFIID, and when bound, TAF1 becomes a histone acetyltransferase (HAT), further acetylating adjacent H3 and H4 histones and recruiting more HATs in the process.[11] DNA wraps around the histones of chromatin, and the acetylation of these histone tails leads to reduction of the positive charge associated with the histones. This results in the negatively-charged DNA to lose affinity for the histone, allowing for more space for transcription factors to bind promoter regions and further facilitate expression.
Eventually, the processes of histone acetylation and consequent chromatin remodeling allow for greater expression of target genes, including those involved in adult neurogenesis. The most prominently studied and well-understood regulators of chromatin remodeling, which play an important role in adult neurogenesis are histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs add acetyl groups to nucleosomes, while HDACs remove them. The acetylation of histones leads to decreased condensation of the nucleosomes to target DNA, and increases the likelihood that gene expression may occur by freeing up the DNA targets to bind to their respective transcriptional factors. This process is involved in neural proliferation regulation, as different neuronal cell genes are expressed and repressed. Deacetylation of histones leads to the reverse, and increases the likelihood for the repression of gene expression.[12]
Role of histone modifications in adult neurogenesis
HDAC inhibitors (HDACi), such as valproic acid (VPA) and trichostatin A can promote proliferation of adult neurogenesis through the inhibition of HDAC activity, inducing differentiation of adult progenitor cells.[12] Neural-expressed HDACs interact with Tlx, an essential neural stem cell regulator, to suppress TLX target genes. This includes the cyclin-dependent kinase inhibitor P21 and the tumor suppressor gene Pten to promote neural stem cell proliferation.[1] Inhibition of HDACs by the antiepileptic drug valproic acid induces neuronal differentiation as in embryonic neurogenesis, but also inhibits glial cell differentiation of adult neural stem cells. This is likely mediated through upregulation of neuronal specific genes such as the neurogeneic basic helix-loop-helix transcription factors NEUROD, NEUROGENENIN1, and Math1. Conditional loss of HDAC1 and HDAC2 in neural progenitor cells prevented them from differentiating into neurons and their loss in oligodendrytic progenitor cells disrupted oligodendrocyte formations, suggesting that histone deacetlyation plays important but varying roles in different stages of neuronal development.[1]
miRNAs
MicroRNAs (miRNAs) are small noncoding RNAs that play a significant role in eukaryotic epigenetic regulation. miRNAs function to modulate protein expression levels of their mRNA targets without affecting the sequences of the genes of interest. While miRNAs play a large role in the modulation of epigenetic mechanisms, they are also modified and regulated by other epigenetic factors including DNA methylation, histone modifications and other RNA modifications.[13] Together, miRNAs create an epigenetic feedback loop with other epigenetic factors to affect the expression levels of specific genes. A number of specific miRNAs have been implicated as agents of epigenetic regulation in adult neurogenesis. miR-9 targets the nuclear receptor TLX in adult neurogenesis to promote neural differentiation and inhibit neural stem cell proliferation. It also influences neuronal subtype specification and regulates axonal growth, branching, and targeting in the central nervous system through interactions with HES1, a neural stem cell homeostasis molecule. miR-124 promotes cell cycle exit and neuronal differentiation in adult neurogenesis. Mouse studies have shown that ectopic expression of miR-124 showed premature neural progenitor cell differentiation and exhaustion in the subventricular zone.
In addition to miR-9 and miR-124, other miRNAs play essential roles in regulation of adult neurogenesis. miR-137, miR-184 and miR-195 regulate adult neural stem cell proliferation, with their over-expression leading to up-regulated proliferation while their down-regulation leads to a decrease in neuronal proliferation.[14] Methyl-CpG binding protein 1 (MBD1) represses miR-184, which is a microRNA responsible for proliferation of adult neural stem/progenitor cells (aNSCs) along with the inhibition of differentiating these cells. miR-184 regulates embryonic brain development by binding to the mRNA for the Numblike (Numbl) protein and altering its expression. MBD1, Numbl, and miR-184 all work together to regulate the proliferation and differentiation of aNSCs.[15] In addition, miR-195 works closely with MBD1 to regulate aNSC proliferation and differentiation. mIR-194 and MBD1 form a negative regulatory loop in aNSCs and work to repress the expression of each other. Inhibition of miR-195 promotes aNSC differentiation. Once differentiation has occurred, levels of miR-195 decreases.[16]
Astrocyte reprogramming
Astrocytes are glial cells that forms the blood brain barrier, support synapses as well as guide axons.[17] Unlike neurons, these specialized glial cells are able to alter their cell fate prior to reaching full maturation and “dedifferentiate,” in large part due to epigenetic factors. This dedifferentiation allows astrocytes to potentially reach a different cell fate entirely, so long as this dedifferentiation occurs before complete maturation occurs, and can lead to their consequent differentiation and conversion from glial cells into neurons in the adult brain.[18] Prior to complete maturation while dedifferentiation is still possible, the expression of genes Mash1, NeuroG1 and NeuroG2 can reprogram astrocytes into neurons.[19] In addition to the expression of these genes in brain cells, multiple epigenetic factors play a role in these gene's expression patterns. An up-regulation of acetylation at H3K9 and H3K14 residues adjacent to both NeuroG1 and NeuroG2 genes has been shown to accompany astrocyte dedifferentiation, over-expression and/or forced expression of these genes can directly induce the differentiation of astrocytes.
In addition, silencing of methylation mechanisms, specifically the silencing of many classes of DNA methyltransferases, which themselves are involved in silencing expression, inhibits the progenitor cells of astrocytes from differentiating back to their original fate as glial cells.[20] Despite this knowledge of the mechanism of methylation repression, the identity of these silenced genes is not yet fully known. While this overall repression of methylation is necessary to prevent expression of specific genes needed to allow an astrocyte to fully mature and reach an astrocytic cell fate, it was found that the over-expression of one specific histone methyltransferase, Ezh2, which catalyzes the tri-methylation of H3K27, represses genes needed for astrocyte maintenance, thus allowing the cell to retain its neural stem cell morphology. This demonstrates that differential methylation by distinct methyltransferases and their consequent repression or over-expression have differing roles in the dedifferentiation of astrocytes to form neurons. Furthermore, while not sufficient to induce astrocyte dedifferentiation alone, Ezh2 is necessary for astrocytes to dedifferentiate as they are inhibited from reaching complete maturity into their original cell fate. Once in this inhibited stage, expression of the gene NeuroD4 in these specified glial cells has been shown to lead to neuronal formation, and thus neurogenesis, from the dedifferentiated astrocytes in adult mammalian brains.[18]
In memory
The Growth Arrest and DNA Damage inducible 45 (Gadd45) gene family plays a large role in the hippocampus. Gadd45 facilitates hippocampal long-term potentiation and enhances persisting memory for motor performance, aversive conditioning, and spatial navigation.[21] Additionally, DNA methylation has been shown to be important for activity-dependent modulation of adult neurogenesis in the hippocampus, which is mediated by GADD45b. GADD45b seems to act as a sensor in mature neurons for environmental changes which it expresses through these methylation changes.[1] This was determined by examining the effects of applying an electric stimulus to the hippocampal dentate gyrus (DG) in normal and GADD45b knockout mice. In normal mice application of electrical stimulation to the DG increased neurogenesis by increasing BDNF. However, in GADD45b deficient mice the electrical stimulus had less of an effect. Further examination revealed that around 1.4% of CpG islands in DG neurons are actively methylated and demethylated upon electric shock. This shows that the post-mitotic methylation states of neurons are not static and given that electric shock equipment such as that used in the study has been shown to have therapeutic effects to human patients with depression and other psychiatric disorders, the possibility remains that epigenetic mechanisms may play an important role in the pathophysiology of neuropsychiatric disorders.[2][8] DNMT1 and DNMT3a are both required in conjunction for learning, memory, and synaptic plasticity.[8]
Epigenetic dysregulation and neurological disorders
Epigenetic dysregulation, or alterations in epigenomic machinery, can cause DNA methylation and histone acetylation processes to go rogue. The epigenetic machinery influences neural differentiation regulation (i.e. neurogenesis) [22] and are also involved in processes related to memory consolidation and learning in healthy individuals.[23] Increasing age can produce various epigenetic changes such as reduced global heterochromatin, nucleosome remodeling, altered histone marks, and changes in DNA methylation. For instance, nucleosome loss occurs due to aging because core histone proteins are lost and less protein synthesis occurs.[24] As aging is the main risk for many neurological disorders, epigenetic dysregulation can in turn lead to alterations on the transcriptional level of genes involved in the pathogenesis of neural degenerative diseases such as Parkinson's disease, Alzheimer's disease, Huntington's disease, schizophrenia, and bipolar disease.[1][25]
Alzheimer's disease
MicroRNA expression is critical for neurogenesis. In patients with Alzheimer's disease miR-9 and miR-128 is upregulated, while miR-15a is downregulated.[4] Alzheimer's patients also show decreases in brain-derived neurotrophic factor, which has been shown to be repressed through DNA methylation.[8] Although what has been argued as the most evidence for epigenetic influence in Alzheimer's is the gene which controls the protein responsible for amyloid plaque formation, App. This gene has very high GC content in its promoter region, meaning that it is highly susceptible to DNA methylation. This promoter site has been shown to naturally reduce methylation with aging, exemplifying the parallels between aging and Alzheimer's already well known.[26][27] Heavy metals also seem to interfere with epigenetic mechanisms. Specifically in the case of APP, lead exposure earlier in life has been shown to cause a marked over-expression of the APP protein, leading to more amyloid plaque later in life in the aging brain.[27]
DNA methylation's age relation has been further investigated in the promoter regions of several Alzheimer's related genes in the brains of postmortem late-onset Alzheimer's disease patients. The older patients seem to have more abnormal epigenetic machinery than the younger patients, despite the fact that both had died from Alzheimer's. Though this in of itself is not conclusive evidence of anything, it has led to an age-related epigenetic drift theory where abnormalities in epigenetic machinery and exposure to certain environmental factors which occur earlier in life lead to aberrant DNA methylation patterns far later, contributing to sporadic Alzheimer's Disease predisposition.[27]
Histone modifications may also have an impact in Alzheimer's disease, but the differences between HDAC effects in rodent brains compared to human brains have researchers puzzled.[27] As the focus for neurodegenerative diseases begins to shift towards epigenetic pharmacology, it can be expected that the interactions of histone modifications with respect to neurogenesis will become more clear.
Huntington’s disease
Histone acetylation has received increasing support over the years as a proposed mechanism through which epigenetic dysregulation leads changes in gene expression that contribut to HD.[28] Studies that look at mice with HD versus the wild type (WT) have shown that specific gene loci (Drd2, Penk1, Actb, and Grin1) decrease in histone acetylation levels, suggesting that a mutation of the Huntington (HTT) gene and its overexpression may be the cause of this epigenetic dysregulation.
It has been thought that HDAC inhibitors (HDACi's) could partially reverse the low acetylation levels seen in patients with HD. Preclinical studies have been performed using various HDACi's [such as suberoxylanilide hydroxamic acid (SAHA), Trichostatin A (TSA), phenylbutyrate, and sodium butyrate (NaB)] that target HDACI and HDACII. Although these inhibitors improve some phenotypes of HD in mice, such as neuropathology and motor function, these beneficial effects do not lead towards a conclusion for the definitive need for increasing acetylation levels in HD patients. However, inactivation of a target of SAHA, Hdac 4, alleviates neurodegenerative complications in mice with HD through a transcription-independent mechanism which acts upon mutant Htt aggregation processes-which may indicate that there is a mechanism involving non-histone proteins.[29] The proposed mechanism through which SAHA is speculated to act is through a RANBP2-mediated proteasome degradation model-which likely comes about as a general outcome of HDAC inhibitor actions. In this mechanism, SAHA is shown to down-regulate Hdac 4 through an increase in sumoylation, which is then followed up with the activation of degradation through a proteasomal pathway. This mechanism reveals the connectivity between acetylation, deacetylation, and sumoylation processes.[30]
As of 2014, HDACi treatment has not been shown to restore normal expression of neuronal-identity genes.[31] However clinical studies using HDACi are currently ongoing and the results are pending, with the Phase II studies showing promise for safe and tolerable use of several compounds such as phenylbutyrate.
Non-histone-mediated beneficial effects of HDACi have also been documented in models of Parkinson disease, suggesting common mechanisms between several neurodegenerative diseases.
Parkinson’s disease
DNA methylation analysis showed that there is significant dysregulation of methylation on CpG islands in patients with PD when compared to healthy individuals. Although this was genome-wide, this also occurred on many PD risk genes.[32]
Mitochondrial DNA methylation of cytosine has also been shown to fluctuate over time due to age variance, as there is a growing body of literature linking mtDNA methylation to aging and oxidative stress.[33] A study from 2015 by Hashizume et al. showed that SHMT2 mRNA levels are significantly reduced in the fibroblasts of old people when compared to younger individuals. The study also further indicated that decreased GCAT and SHMT2 levels of gene expression via shRNA and siRNA, respectively, in the fibroblasts of young patients led to a respiratory chain dysfunction typical for senile individuals-suggesting that an epigenetic mechanism may be the cause for the phenotypic change. As mitochondria plays a role in the development of the PD,[34] further research into the area will help uncover any implications that mitochondrial DNA methylation plays in the pathogenesis of PD.
The use of dopaminergic neurons that have been isolated from the PD patients indicated that there were increases in acetylation (at H2A, H3 and H4) when compared to the age-control group.[32] Another study involving MPP+ (a compound that can cause a disease state resembling mammals and humans with PD[35])-treated cells and (MPP+)-treated mouse brains showed decreased HDAC levels, as well as in midbrain samples from patients with PD. This is seen potentially due to how MPP+ promotes the breakdown of HDAC1 and HDAC2 via autophagy, a bodily process of cycling out old cells to make room for newer, healthier cells.[36] These results point towards the stress of histone modifications in regards to chromatin remodeling and its implication in the pathogenesis of PD.
miRNAs are also emerging as relevant contributors to neurodegeneration in PD. In particular, the frontal cortex PD patients have shown higher levels of LRRK2 and lower levels of miR-205 when compared to healthy individuals. Connecting this to the findings of miR-205's ability to bind to the 3′ UTR of LRRK2 mRNA and suppress expression, as well as miR-205's prevention of defects after introduction to a R1441G LRRK2 mutation, these results point towards miR-205 and its regulatory role in LRRK2 expression-which in turn suggest a regulatory role in the pathogenesis of PD.
In another study in which increasing microtubule acetylation using deacetylase inhibitors or the tubulin acetylase αTAT1 showed prevention of the association of mutant LRRK2 with microtubules, inhibition of deacetylases HDAC6 and Sirt2 through knockdown processes rescued both axonal transport and locomotor behavior.[37] This further connects to the common mechanisms involving HDACi in various neurodegenerative diseases.
Bipolar Disorder
Bipolar disorders are both highly complex and heritable, which makes it an interesting disorder to examine for epigenetic modifications. DNA methylation, DNA hydroxymethylation, and histone modifications are all capable of contributing to the formation of bipolar disorder.
For example, studies of monozygotic twins revealed that individuals with bipolar disorder had lower methylation of the peptidylprolyl isomerase E-like (PPIEL) gene, which can be attributed to the dopamine transmission. The studies indicated that hypermethylation of SLC6A4, a serotonin transporter gene, is also involved with bipolar disorder. Greater expression of DNA methyltransferase 1 in cortical GABAergic interneurons may enable hypermethylation. Hypermethylation may prompt hydroxymethylation to occur in order to overcompensate for the repressive effects of hypermethylation. The methylation of CpG regions are relevant to bipolar disorders. Patients with bipolar disorder showed lower methylation levels for the CpG region of the KCNQ3 gene, which is responsible for the voltage-gated K+ channel. Childhood maltreatment contributed to the methylation status of CpG2 III of 5-hydroxytryptamine 3A, which alters how maltreatment affects bipolar disorder.
Moreover, therapeutic interventions such as engineered transcription factors could modify chromatin structure to address the epigenetic changes found in those with bipolar disorder. DNA methyltransferase (DNMT) inhibitors and histone deacetylase (HDAC) inhibitors could possibly reverse epigenetic modifications in order to therapeutically address bipolar disorder. DNMT inhibitors and HDAC often produces antidepressant-like effects.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Hu XL, Wang Y, Shen Q (April 2012). "Epigenetic control on cell fate choice in neural stem cells". Protein & Cell. 3 (4): 278–290. doi:10.1007/s13238-012-2916-6. PMC 4729703. PMID 22549586.
- 1 2 3 4 Faigle R, Song H (February 2013). "Signaling mechanisms regulating adult neural stem cells and neurogenesis". Biochimica et Biophysica Acta (BBA) - General Subjects. 1830 (2): 2435–2448. doi:10.1016/j.bbagen.2012.09.002. PMC 3541438. PMID 22982587.
- 1 2 3 4 5 6 7 MuhChyi C, Juliandi B, Matsuda T, Nakashima K (October 2013). "Epigenetic regulation of neural stem cell fate during corticogenesis". International Journal of Developmental Neuroscience. 31 (6): 424–433. doi:10.1016/j.ijdevneu.2013.02.006. PMID 23466416. S2CID 205242753.
- 1 2 3 4 Ji F, Lv X, Jiao J (February 2013). "The role of microRNAs in neural stem cells and neurogenesis". Journal of Genetics and Genomics = Yi Chuan Xue Bao. 40 (2): 61–66. doi:10.1016/j.jgg.2012.12.008. PMID 23439404.
- 1 2 3 4 Sun AX, Crabtree GR, Yoo AS (April 2013). "MicroRNAs: regulators of neuronal fate". Current Opinion in Cell Biology. 25 (2): 215–221. doi:10.1016/j.ceb.2012.12.007. PMC 3836262. PMID 23374323.
- ↑ De Pietri Tonelli D, Pulvers JN, Haffner C, Murchison EP, Hannon GJ, Huttner WB (December 2008). "miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex". Development. 135 (23): 3911–3921. doi:10.1242/dev.025080. PMC 2798592. PMID 18997113.
- ↑ Kawase-Koga Y, Low R, Otaegi G, Pollock A, Deng H, Eisenhaber F, et al. (February 2010). "RNAase-III enzyme Dicer maintains signaling pathways for differentiation and survival in mouse cortical neural stem cells". Journal of Cell Science. 123 (Pt 4): 586–594. doi:10.1242/jcs.059659. PMC 2818196. PMID 20103535.
- 1 2 3 4 5 Lv J, Xin Y, Zhou W, Qiu Z (July 2013). "The epigenetic switches for neural development and psychiatric disorders". Journal of Genetics and Genomics = Yi Chuan Xue Bao. 40 (7): 339–346. doi:10.1016/j.jgg.2013.04.007. PMID 23876774.
- ↑ Pepenella S, Murphy KJ, Hayes JJ (September 2014). "A distinct switch in interactions of the histone H4 tail domain upon salt-dependent folding of nucleosome arrays". The Journal of Biological Chemistry. 289 (39): 27342–27351. doi:10.1074/jbc.M114.595140. PMC 4175364. PMID 25122771.
- ↑ Peterson CL (April 2002). "Chromatin remodeling enzymes: taming the machines. Third in review series on chromatin dynamics". EMBO Reports. 3 (4): 319–322. doi:10.1093/embo-reports/kvf075. PMC 1084063. PMID 11943761.
- ↑ Hilton TL, Li Y, Dunphy EL, Wang EH (May 2005). "TAF1 histone acetyltransferase activity in Sp1 activation of the cyclin D1 promoter". Molecular and Cellular Biology. 25 (10): 4321–4332. doi:10.1128/MCB.25.10.4321-4332.2005. PMC 1087727. PMID 15870300.
- 1 2 Hsieh J, Zhao X (June 2016). "Genetics and Epigenetics in Adult Neurogenesis". Cold Spring Harbor Perspectives in Biology. 8 (6): a018911. doi:10.1101/cshperspect.a018911. PMC 4888816. PMID 27143699.
- ↑ Yao Q, Chen Y, Zhou X (August 2019). "The roles of microRNAs in epigenetic regulation". Current Opinion in Chemical Biology. 51: 11–17. doi:10.1016/j.cbpa.2019.01.024. PMID 30825741. S2CID 73490246.
- ↑ Szulwach KE, Li X, Smrt RD, Li Y, Luo Y, Lin L, et al. (April 2010). "Cross talk between microRNA and epigenetic regulation in adult neurogenesis". The Journal of Cell Biology. 189 (1): 127–141. doi:10.1083/jcb.200908151. PMC 2854370. PMID 20368621.
- ↑ Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, Zhao X (May 2010). "Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation". Cell Stem Cell. 6 (5): 433–444. doi:10.1016/j.stem.2010.02.017. PMC 2867837. PMID 20452318.
- ↑ Liu C, Teng ZQ, McQuate AL, Jobe EM, Christ CC, von Hoyningen-Huene SJ, et al. (2013-01-17). Van Wijnen A (ed.). "An epigenetic feedback regulatory loop involving microRNA-195 and MBD1 governs neural stem cell differentiation". PLOS ONE. 8 (1): e51436. Bibcode:2013PLoSO...851436L. doi:10.1371/journal.pone.0051436. PMC 3547917. PMID 23349673.
- ↑ Blackburn D, Sargsyan S, Monk PN, Shaw PJ (September 2009). "Astrocyte function and role in motor neuron disease: a future therapeutic target?". Glia. 57 (12): 1251–1264. doi:10.1002/glia.20848. PMID 19373940. S2CID 205833357.
- 1 2 Griffiths BB, Bhutani A, Stary CM (June 2020). "Adult neurogenesis from reprogrammed astrocytes". Neural Regeneration Research. 15 (6): 973–979. doi:10.4103/1673-5374.270292. PMC 7034263. PMID 31823866.
- ↑ Berninger B, Costa MR, Koch U, Schroeder T, Sutor B, Grothe B, Götz M (August 2007). "Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia". The Journal of Neuroscience. 27 (32): 8654–8664. doi:10.1523/JNEUROSCI.1615-07.2007. PMC 6672931. PMID 17687043.
- ↑ Bulstrode H, Johnstone E, Marques-Torrejon MA, Ferguson KM, Bressan RB, Blin C, et al. (April 2017). "Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators". Genes & Development. 31 (8): 757–773. doi:10.1101/gad.293027.116. PMC 5435889. PMID 28465359.
- ↑ Sultan FA, Wang J, Tront J, Liebermann DA, Sweatt JD (November 2012). "Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity". The Journal of Neuroscience. 32 (48): 17059–17066. doi:10.1523/JNEUROSCI.1747-12.2012. PMC 3518911. PMID 23197699.
- ↑ Yao B, Christian KM, He C, Jin P, Ming GL, Song H (September 2016). "Epigenetic mechanisms in neurogenesis". Nature Reviews. Neuroscience. 17 (9): 537–549. doi:10.1038/nrn.2016.70. PMC 5610421. PMID 27334043.
- ↑ Delgado-Morales R, Agís-Balboa RC, Esteller M, Berdasco M (2017-06-29). "Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders". Clinical Epigenetics. 9 (1): 67. doi:10.1186/s13148-017-0365-z. PMC 5493012. PMID 28670349.
- ↑ Kane AE, Sinclair DA (February 2019). "Epigenetic changes during aging and their reprogramming potential". Critical Reviews in Biochemistry and Molecular Biology. 54 (1): 61–83. doi:10.1080/10409238.2019.1570075. PMC 6424622. PMID 30822165.
- ↑ Dempster EL, Pidsley R, Schalkwyk LC, Owens S, Georgiades A, Kane F, et al. (December 2011). "Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder". Human Molecular Genetics. 20 (24): 4786–4796. doi:10.1093/hmg/ddr416. PMC 3221539. PMID 21908516.
- ↑ Balazs R, Vernon J, Hardy J (July 2011). "Epigenetic mechanisms in Alzheimer's disease: progress but much to do". Neurobiology of Aging. 32 (7): 1181–1187. doi:10.1016/j.neurobiolaging.2011.02.024. PMID 21669333. S2CID 20303624.
- 1 2 3 4 Daniilidou M, Koutroumani M, Tsolaki M (2011). "Epigenetic mechanisms in Alzheimer's disease". Current Medicinal Chemistry. 18 (12): 1751–1756. doi:10.2174/092986711795496872. PMID 21466476.
- ↑ Francelle L, Lotz C, Outeiro T, Brouillet E, Merienne K (2017-01-30). "Contribution of Neuroepigenetics to Huntington's Disease". Frontiers in Human Neuroscience. 11: 17. doi:10.3389/fnhum.2017.00017. PMC 5276857. PMID 28194101.
- ↑ Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP (2011-11-28). "SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease". PLOS ONE. 6 (11): e27746. Bibcode:2011PLoSO...627746M. doi:10.1371/journal.pone.0027746. PMC 3225376. PMID 22140466.
- ↑ Scognamiglio A, Nebbioso A, Manzo F, Valente S, Mai A, Altucci L (October 2008). "HDAC-class II specific inhibition involves HDAC proteasome-dependent degradation mediated by RANBP2". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1783 (10): 2030–2038. doi:10.1016/j.bbamcr.2008.07.007. PMID 18691615.
- ↑ Coppedè F (2014). "The potential of epigenetic therapies in neurodegenerative diseases". Frontiers in Genetics. 5: 220. doi:10.3389/fgene.2014.00220. PMC 4094885. PMID 25071843.
- 1 2 Pavlou MA, Outeiro TF (2017). "Epigenetics in Parkinson's Disease". In Delgado-Morales R (ed.). Neuroepigenomics in Aging and Disease. Advances in Experimental Medicine and Biology. Vol. 978. Cham: Springer International Publishing. pp. 363–390. doi:10.1007/978-3-319-53889-1_19. ISBN 978-3-319-53889-1. PMID 28523556.
- ↑ Zinovkina LA, Zinovkin RA (December 2015). "DNA Methylation, Mitochondria, and Programmed Aging". Biochemistry. Biokhimiia. 80 (12): 1571–1577. doi:10.1134/S0006297915120044. PMID 26638681. S2CID 17044164.
- ↑ Mhyre TR, Boyd JT, Hamill RW, Maguire-Zeiss KA (2012). "Parkinson's Disease". Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease. Subcellular Biochemistry. Vol. 65. pp. 389–455. doi:10.1007/978-94-007-5416-4_16. ISBN 978-94-007-5415-7. PMC 4372387. PMID 23225012.
- ↑ Ito K, Eguchi Y, Imagawa Y, Akai S, Mochizuki H, Tsujimoto Y (2017-02-27). "MPP+ induces necrostatin-1- and ferrostatin-1-sensitive necrotic death of neuronal SH-SY5Y cells". Cell Death Discovery. 3: 17013. doi:10.1038/cddiscovery.2017.13. PMC 5327502. PMID 28250973.
- ↑ Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z (February 2016). "Regulation of Histone Acetylation by Autophagy in Parkinson Disease". The Journal of Biological Chemistry. 291 (7): 3531–3540. doi:10.1074/jbc.M115.675488. PMC 4751393. PMID 26699403.
- ↑ Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, et al. (October 2014). "Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations". Nature Communications. 5: 5245. Bibcode:2014NatCo...5.5245G. doi:10.1038/ncomms6245. PMC 4208097. PMID 25316291.
External links
- Database of known microRNAs: http://www.miRbase.org
- Article on histone methylation visualization through fluorescent imaging