| Familial Mediterranean fever | |
|---|---|
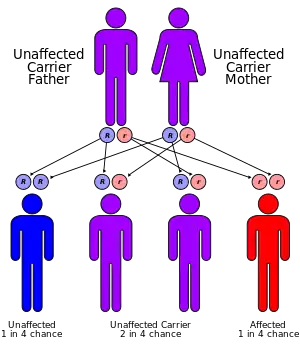 | |
| Familial Mediterranean fever has an autosomal recessive pattern of inheritance | |
| Specialty | Rheumatology, Immunology |
Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder.[1]: 149 FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin.[2] While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews,[3][4] Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.[5][6][7][8]
The disorder has been given various names, including familial paroxysmal polyserositis, periodic peritonitis, recurrent polyserositis, benign paroxysmal peritonitis, periodic disease or periodic fever, Reimann periodic disease or Reimann syndrome, Siegal-Cattan-Mamou disease, and Wolff periodic disease.[9][10][11] Note that "periodic fever" can also refer to any of the periodic fever syndromes.
Signs and symptoms
Attacks
There are seven types of attacks. Ninety percent of all patients have their first attack before they are 18 years old. All develop over 2–4 hours and last anywhere from 6 hours to 5 days. Most attacks involve fever.[12]
- Abdominal attacks, featuring abdominal pain, affect the whole abdomen with all signs of peritonitis (inflammation of abdominal lining), and acute abdominal pain like appendicitis. They occur in 95% of all patients and may lead to unnecessary laparotomy. Incomplete attacks, with local tenderness and normal blood tests, have been reported.
- Joint attacks mainly occur in large joints, especially in the legs. Usually, only one joint is affected. 75% of all FMF patients experience joint attacks.
- Chest attacks include pleuritis (inflammation of the pleura) and pericarditis (inflammation of the pericardium). Pleuritis occurs in 40% of patients and makes it difficult to breathe or lie flat, but pericarditis is rare.
- Scrotal attacks due to inflammation of the tunica vaginalis are somewhat rare but may be mistaken for testicular torsion.
- Myalgia (rare in isolation)
- Erysipeloid rashes (a skin reaction on the legs that can mimic cellulitis, rare in isolation)
 Erysipeloid rashes in Familial Mediterranean Fever
Erysipeloid rashes in Familial Mediterranean Fever
Diagnostic criteria
Various diagnostic criteria have been set, but the Tel-Hashomer clinical criteria is widely recognized for the diagnosis of FMF. It has more than 95% and 97% sensitivity and specificity, respectively.[13]
For the criteria, typical attacks consist of all the following: recurrent (three or more episodes), febrile (rectal ≥ 38 °C), painful inflammation, and a short duration of 12 to 72 hours.
Incomplete attacks (must be recurrent) are differing from typical attacks in at least one feature as follows: temperature <38 °C, attack duration longer or shorter than specified (but no less than 6 hours and no more than 7 days), localized abdominal attacks, no signs of peritonitis during the attacks, and arthritis in a location other than the hip, knee or ankle.
Complications
AA-amyloidosis with kidney failure is a complication and may develop without overt crises. AA amyloid protein is produced in very large quantities during attacks, and at a low rate between them, and accumulates mainly in the kidney, as well as the heart, spleen, gastrointestinal tract, and thyroid.[12]
There appears to be an increase in the risk for developing particular vasculitis-related diseases[14] (e.g. Henoch–Schönlein purpura, Polyarteritis nodosa, and Behçet's disease), spondylarthropathy, prolonged arthritis of certain joints and protracted myalgia.[12]
Genetics
The MEFV gene is located on the short arm of chromosome 16 (16p13). Many different mutations of the MEFV gene can cause the disorder. Having one mutation is unlikely to cause the condition. Having two mutations (either a copy from both parents, or two different mutations with one from each parent) is the threshold for a genetic diagnosis of FMF. However, most individuals who comply with the genetic diagnosis of FMF remain asymptomatic or undiagnosed. Whether this is due to modifier genes or environmental factors remains to be established.[4]
Pathophysiology
Virtually all cases are due to a mutation in the Mediterranean Fever (MEFV) gene on the chromosome 16, which codes for a protein called pyrin or marenostrin. Various mutations of this gene lead to FMF, although some mutations cause a more severe picture than others. Mutations occur mainly in exons 2, 3, 5 and 10.[12]
The function of pyrin has not been completely elucidated, but in short, it is a protein that binds to the adaptor ASC and the pro form of the enzyme caspase-1 to generate multiprotein complexes called inflammasomes in response to certain infections. In healthy individuals, pyrin-mediated inflammasome assembly (which leads to the caspase 1) dependent processing and secretion of the pro-inflammatory cytokines (such as interleukin-18 (IL-18) and IL-1β) is a response to enterotoxins from certain bacteria.[15] The gain-of-function mutations in the MEFV gene render Pyrin hyperactive, and subsequently, the formation of the inflammasomes becomes more frequent.[16]
The pathophysiology of familial Mediterranean fever has recently undergone significant advances: at basal state, pyrin is kept inactive by a chaperone protein (belonging to the family of 14.3.3 proteins) linked to pyrin through phosphorylated serine residues.[17][18] The dephosphoration of pyrin is an essential prerequisite for the activation of the pyrin inflammasome. Inactivation of RhoA GTPases (by bacterial toxins, for example) leads to the inactivation of PKN1 / PKN2 kinases and dephosphoration of pyrin.[19] In healthy subjects, the dephosphorylation step alone does not cause activation of the pyrin inflammasome. In contrast, in FMF patients, the dephosphorylation of serines is sufficient to trigger the activation of the pyrin inflammasome.[20] This suggests that there is a two-level regulation and that the second regulatory mechanism (independent of (de)phosphorylation) is deficient in FMF patients. This deficient mechanism is probably located at the level of the B30.2 domain (exon 10) where most of the pathogenic mutations associated with FMF are located. It is probably the interaction of this domain with the cytoskeleton (microtubules) that is failing, as suggested by the efficacy of colchicine.[21]
It is not conclusively known what exactly sets off the attacks, and why overproduction of IL-1 would lead to particular symptoms in particular organs (e.g. joints or the peritoneal cavity).[12] However, steroid hormone catabolites (pregnanolone and etiocholanolone) have been shown to activate the pyrin inflammasome, in vitro, by interacting with the B30.2 domain (coded by exon 10).[22]
Diagnosis
The diagnosis is clinically made on the basis of the history of typical attacks, especially in patients from the ethnic groups in which FMF is more highly prevalent. An acute phase response is present during attacks, with high C-reactive protein levels, an elevated white blood cell count and other markers of inflammation. In patients with a long history of attacks, monitoring the kidney function is of importance in predicting chronic kidney failure.[12]
A genetic test is also available to detect mutations in the MEFV gene. Sequencing of exons 2, 3, 5, and 10 of this gene detects an estimated 97% of all known mutations.[12]
A specific and highly sensitive test for FMF is the "metaraminol provocative test (MPT)", whereby a single 10 mg infusion of metaraminol is administered to the patient. A positive diagnosis is made if the patient presents with a typical, albeit milder, FMF attack within 48 hours. As MPT is more specific than sensitive, it does not identify all cases of FMF, although a positive MPT can be very useful.[23][24]
Treatment
Attacks are self-limiting, and require analgesia and NSAIDs (such as diclofenac).[12] Colchicine, a drug otherwise mainly used in gout, decreases attack frequency in FMF patients. The exact way in which colchicine suppresses attacks is unclear. While this agent is not without side effects (such as abdominal pain and muscle pains), it may markedly improve quality of life in patients. The dosage is typically 1–2 mg a day. Development of amyloidosis is delayed with colchicine treatment. Interferon is being studied as a therapeutic modality.[12] Some advise discontinuation of colchicine before and during pregnancy, but the data are inconsistent, and others feel it is safe to take colchicine during pregnancy.[25]
Approximately 5–10% of FMF cases are resistant to colchicine therapy alone. In these cases, adding anakinra to the daily colchicine regimen has been successful.[26] Canakinumab, an anti-interleukin-1-beta monoclonal antibody, has likewise been shown to be effective in controlling and preventing flare-ups in patients with colchicine-resistant FMF and in two additional autoinflammatory recurrent fever syndromes: mevolonate kinase deficiency (hyper-immunoglobulin D syndrome, or HIDS) and tumor necrosis factor receptor-associated periodic syndrome (TRAPS).[27]
Epidemiology
FMF affects groups of people originating from around the Levant or Eastern Mediterranean (hence its name); it is thus most prominent among those from or with ancestry from the regions including Arabs, Armenians, Jews (particularly Sephardi, Mizrahi, and to a lesser degree Ashkenazi Jews), and Turks.[3][12][28][29]
History
A New York City allergist, Sheppard Siegal, first described the attacks of peritonitis in 1945; he termed this "benign paroxysmal peritonitis", as the disease course was essentially benign.[30] Dr Hobart Reimann, working in the American University in Beirut, described a more complete picture which he termed "periodic disease".[31][32] French physicians Henry Mamou and Roger Cattan described the complete disease with renal complications in 1952.[33][34]
See also
References
- ↑ James W, Berger T, Elston D (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ Chae JJ, Wood G, Richard K, Jaffe H, Colburn NT, Masters SL, et al. (September 2008). "The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment". Blood. 112 (5): 1794–803. doi:10.1182/blood-2008-01-134932. PMC 2518886. PMID 18577712.
- 1 2 Stoffman N, Magal N, Shohat T, Lotan R, Koman S, Oron A, et al. (April 2000). "Higher than expected carrier rates for familial Mediterranean fever in various Jewish ethnic groups". European Journal of Human Genetics. 8 (4): 307–10. doi:10.1038/sj.ejhg.5200446. PMID 10854115.
- 1 2 Gershoni-Baruch R, Shinawi M, Leah K, Badarnah K, Brik R (August 2001). "Familial Mediterranean fever: prevalence, penetrance and genetic drift". European Journal of Human Genetics. 9 (8): 634–7. doi:10.1038/sj.ejhg.5200672. PMID 11528510.
- ↑ "Familial Mediterranean fever". Mayo Clinic.
- ↑ Papadopoulos V, Mitroulis I, Giaglis S (January 2010). "MEFV heterogeneity in Turkish Familial Mediterranean Fever patients". Molecular Biology Reports. 37 (1): 355–8. doi:10.1007/s11033-009-9779-9. PMID 19714479. S2CID 7306747.
- ↑ Saeed D, Mortaza B, Tooba M (15 December 2010). "The Prevalence of Genetic Disorders in East Azerbaijan Province". Urmia Medical Journal. 21 (4): 339–346.
- ↑ Stoffman N, Magal N, Shohat T, Lotan R, Koman S, Oron A, et al. (April 2000). "Higher than expected carrier rates for familial Mediterranean fever in various Jewish ethnic groups" (PDF). European Journal of Human Genetics. 8 (4): 307–10. doi:10.1038/sj.ejhg.5200446. PMID 10854115.
- ↑ Dugdale III DC, Vyas J (2010-09-15). "Familial Mediterranean fever - PubMed Health". PubMed Health. National Centre for Biotechnology Information. Archived from the original on 2012-09-10. Retrieved 2011-04-24.
- ↑ "Siegal-Cattan-Mamou syndrome". Retrieved February 19, 2021.
- ↑ "Familial Mediterranean fever - Genetics Home Reference". Genetics Home Reference. U.S. National Library of Medicine. 2011-04-14. Archived from the original on 2011-06-05. Retrieved 2011-04-24.
- 1 2 3 4 5 6 7 8 9 10 Livneh A, Langevitz P (September 2000). "Diagnostic and treatment concerns in familial Mediterranean fever". Baillière's Best Practice & Research. Clinical Rheumatology. 14 (3): 477–98. doi:10.1053/berh.2000.0089. PMID 10985982.
- ↑ Livneh, Avi; Langevitz, Pnina; Zemer, Deborah; Zaks, Nurit; Kees, Salim; Lidar, Tzvi; Migdal, Amiel; Padeh, Shai; Pras, Mordechai (1997). "Criteria for the diagnosis of familial mediterranean fever". Arthritis & Rheumatism. 40 (10): 1879–1885. doi:10.1002/art.1780401023. PMID 9336425.
- ↑ Yalçınkaya, Fatoş; Özçakar, Z. Bı̇rsı̇n; Kasapçopur, Özgür; Öztürk, Ayşenur; Akar, Nejat; Bakkaloğlu, Ayşı̇n; Arısoy, Nı̇l; Ekı̇m, Mesı̇ha; Özen, Seza (December 2007). "Prevalence of the MEFV Gene Mutations in Childhood Polyarteritis Nodosa". The Journal of Pediatrics. 151 (6): 675–678. doi:10.1016/j.jpeds.2007.04.062. PMID 18035151.
- ↑ Alexandra A. Mushegian (2016). "An alternative path for pyrin". Science Signaling. 9 (459): ec299. doi:10.1126/scisignal.aam6054. S2CID 51606430.
- ↑ Ratner D (May 11, 2016). Activation and Inhibition of Multiple Inflammasome Pathways by the Yersinia Pestis Type Three Secretion System (Ph.D. thesis). University of Massachusetts Medical School.
- ↑ Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. (May 2011). "Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice". Immunity. 34 (5): 755–68. doi:10.1016/j.immuni.2011.02.020. PMC 3129608. PMID 21600797.
- ↑ Jamilloux Y, Magnotti F, Belot A, Henry T (April 2018). "The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes". Pathogens and Disease. 76 (3). doi:10.1093/femspd/fty020. PMID 29718184.
- ↑ Park YH, Wood G, Kastner DL, Chae JJ (August 2016). "Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS". Nature Immunology. 17 (8): 914–21. doi:10.1038/ni.3457. PMC 4955684. PMID 27270401.
- ↑ Magnotti F, Lefeuvre L, Benezech S, Malsot T, Waeckel L, Martin A, et al. (November 2019). "Pyrin dephosphorylation is sufficient to trigger inflammasome activation in familial Mediterranean fever patients". EMBO Molecular Medicine. 11 (11): e10547. doi:10.15252/emmm.201910547. PMC 6835204. PMID 31589380.
- ↑ Van Gorp H, Saavedra PH, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, et al. (December 2016). "Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation". Proceedings of the National Academy of Sciences of the United States of America. 113 (50): 14384–14389. Bibcode:2016PNAS..11314384V. doi:10.1073/pnas.1613156113. PMC 5167202. PMID 27911804.
- ↑ Magnotti, Flora; Chirita, Daria; Dalmon, Sarah; Martin, Amandine; Bronnec, Pauline; Sousa, Jeremy; Helynck, Olivier; Lee, Wonyong; Kastner, Daniel L.; Chae, Jae Jin; McDermott, Michael F.; Belot, Alexandre; Popoff, Michel; Sève, Pascal; Georgin-Lavialle, Sophie; Munier-Lehmann, Hélène; Tran, Tu Anh; De Langhe, Ellen; Wouters, Carine; Jamilloux, Yvan; Henry, Thomas (2022). "Steroid hormone catabolites activate the pyrin inflammasome through a non-canonical mechanism". Cell Reports. 41 (2): 111472. doi:10.1016/j.celrep.2022.111472. PMC 9626387. PMID 36223753.
- ↑ Barakat MH, El-Khawad AO, Gumaa KA, El-Sobki NI, Fenech FF (March 1984). "Metaraminol provocative test: a specific diagnostic test for familial Mediterranean fever". Lancet. 1 (8378): 656–7. doi:10.1016/s0140-6736(84)92172-x. PMID 6142351. S2CID 23211155.
- ↑ Huppertz HI, Michels H (May 1988). "[The metaraminol provocation test in the diagnosis of familial Mediterranean fever]". Monatsschrift Kinderheilkunde. 136 (5): 243–5. PMID 3405225.
- ↑ Michael O, Goldman RD, Koren G (August 2003). "Safety of colchicine therapy during pregnancy". Canadian Family Physician. 49: 967–9. PMC 2214270. PMID 12943352. Archived from the original on 2009-01-30.
- ↑ Calligaris L, Marchetti F, Tommasini A, Ventura A (June 2008). "The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever". European Journal of Pediatrics. 167 (6): 695–6. doi:10.1007/s00431-007-0547-3. PMC 2292480. PMID 17588171.
- ↑ De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. (May 2018). "Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes" (PDF). The New England Journal of Medicine. 378 (20): 1908–1919. doi:10.1056/NEJMoa1706314. PMID 29768139.
- ↑ Kucuk A, Gezer IA, Ucar R, Karahan AY (2014). "Familial Mediterranean Fever". Acta Medica. 57 (3): 97–104. doi:10.14712/18059694.2014.47. PMID 25649364.
- ↑ Sinha CK, Davenport M (2010). Handbook of Pediatric Surgery. New York: Springer. p. 192. ISBN 9781848821323.
- ↑ Siegal S (February 1949). "Benign paroxysmal peritonitis". Annals of Internal Medicine. 12 (2): 234–47. doi:10.7326/0003-4819-23-1-1. PMID 18124924.
- ↑ Reimann HA (January 1948). "Periodic disease; a probable syndrome including periodic fever, benign paroxysmal peritonitis, cyclic neutropenia and intermittent arthralgia". Journal of the American Medical Association. 136 (4): 239–44. doi:10.1001/jama.1948.02890210023004. PMID 18920089.
- ↑ synd/2503 at Who Named It?
- ↑ Mamou H, Cattan R (1952). "Semaine Des Hôpitaux de Paris". La Maladie Périodique. N°28: 1062–1070.
- ↑ Adwan MH (September 2015). "A brief history of familial Mediterranean fever". Saudi Medical Journal. 36 (9): 1126–7. doi:10.15537/smj.2015.9.12219. PMC 4613641. PMID 26318474.
External links
- Proteopedia 2wl1 information about the MEFV gene.
- GeneReview/NIH/UW entry on Familial Mediterranean Fever
- Familial Mediterranean Fever (FMF) - US National Institute of Arthritis and Musculoskeletal and Skin Diseases