
The fixation index (FST) is a measure of population differentiation due to genetic structure. It is frequently estimated from genetic polymorphism data, such as single-nucleotide polymorphisms (SNP) or microsatellites. Developed as a special case of Wright's F-statistics, it is one of the most commonly used statistics in population genetics. Its values range from 0 to 1, with 0.15 being substantially differentiated and 1 being complete differentiation.
Interpretation
This comparison of genetic variability within and between populations is frequently used in applied population genetics. The values range from 0 to 1. A zero value implies complete panmixis; that is, that the two populations are interbreeding freely. A value of one implies that all genetic variation is explained by the population structure, and that the two populations do not share any genetic diversity.
For idealized models such as Wright's finite island model, FST can be used to estimate migration rates. Under that model, the migration rate is
- ,

where m is the migration rate per generation, and is the mutation rate per generation.[1]

The interpretation of FST can be difficult when the data analyzed are highly polymorphic. In this case, the probability of identity by descent is very low and FST can have an arbitrarily low upper bound, which might lead to misinterpretation of the data. Also, strictly speaking FST is not a distance in the mathematical sense, as it does not satisfy the triangle inequality.
For populations of plants which clearly belong to the same species, values of FST greater than 15% are considered "great" or "significant" differentiation, while values below 5% are considered "small" or "insignificant" differentiation.[2] Values for mammal populations between subspecies, or closely related species, typical values are of the order of 5% to 20%. FST between the Eurasian and North American populations of the gray wolf were reported at 9.9%, those between the Red wolf and Gray wolf populations at between 17% and 18%. The Eastern wolf, a recently recognized highly admixed "wolf-like species" has values of FST below 10% in comparison with both Eurasian (7.6%) and North American gray wolves (5.7%), with the Red wolf (8.5%), and an even lower value when paired with the Coyote (4.5%).[3]
Definition
Two of the most commonly used definitions for FST at a given locus are based on 1) the variance of allele frequencies among populations, and on 2) the probability of identity by descent.
If is the average frequency of an allele in the total population, is the variance in the frequency of the allele among different subpopulations, weighted by the sizes of the subpopulations, and is the variance of the allelic state in the total population, FST is defined as [4]




Wright's definition illustrates that FST measures the amount of genetic variance that can be explained by population structure. This can also be thought of as the fraction of total diversity that is not a consequence of the average diversity within subpopulations, where diversity is measured by the probability that two randomly selected alleles are different, namely . If the allele frequency in the th population is and the relative size of the th population is , then





Alternatively,[5]

where is the probability of identity by descent of two individuals given that the two individuals are in the same subpopulation, and is the probability that two individuals from the total population are identical by descent. Using this definition, FST can be interpreted as measuring how much closer two individuals from the same subpopulation are, compared to the total population. If the mutation rate is small, this interpretation can be made more explicit by linking the probability of identity by descent to coalescent times: Let T0 and T denote the average time to coalescence for individuals from the same subpopulation and the total population, respectively. Then,



This formulation has the advantage that the expected time to coalescence can easily be estimated from genetic data, which led to the development of various estimators for FST.
Estimation
In practice, none of the quantities used for the definitions can be easily measured. As a consequence, various estimators have been proposed. A particularly simple estimator applicable to DNA sequence data is:[6]

where and represent the average number of pairwise differences between two individuals sampled from different sub-populations () or from the same sub-population (). The average pairwise difference within a population can be calculated as the sum of the pairwise differences divided by the number of pairs. However, this estimator is biased when sample sizes are small or if they vary between populations. Therefore, more elaborate methods are used to compute FST in practice. Two of the most widely used procedures are the estimator by Weir & Cockerham (1984),[7] or performing an Analysis of molecular variance. A list of implementations is available at the end of this article.


FST in humans

FST values depend strongly on the choice of populations. Closely related ethnic groups, such as the Danes vs. the Dutch, or the Portuguese vs. the Spaniards show values significantly below 1%, indistinguishable from panmixia. Within Europe, the most divergent ethnic groups have been found to have values of the order of 7% (Lapps vs. Sardinians).
Larger values are found if highly divergent homogenous groups are compared: the highest such value found was at close to 46%, between Mbuti and Papuans.[8]
A genetic distance of 0.125 implies that kinship between unrelated individuals of the same ancestry relative to the world population is equivalent to kinship between half siblings in a randomly mating population. This also implies that if a human from a given ancestral population has a mixed half-sibling, that human is closer genetically to an unrelated individual of their ancestral population than to their mixed half-sibling. [9]
Autosomal genetic distances based on classical markers
In their study The History and Geography of Human Genes (1994), Cavalli-Sforza, Menozzi and Piazza provide some of the most detailed and comprehensive estimates of genetic distances between human populations, within and across continents. Their initial database contains 76,676 gene frequencies (using 120 blood polymorphisms), corresponding to 6,633 samples in different locations. By culling and pooling such samples, they restrict their analysis to 491 populations.
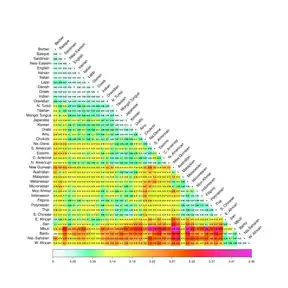
They focus on aboriginal populations that were at their present location at the end of the 15th century when the great European migrations began.[10] When studying genetic difference at the world level, the number is reduced to 42 representative populations, aggregating subpopulations characterized by a high level of genetic similarity. For these 42 populations, Cavalli-Sforza and coauthors report bilateral distances computed from 120 alleles. Among this set of 42 world populations, the greatest genetic distance observed is between Mbuti Pygmies and Papua New Guineans, where the Fst distance is 0.4573, while the smallest genetic distance (0.0021) is between the Danish and the English.
When considering more disaggregated data for 26 European populations, the smallest genetic distance (0.0009) is between the Dutch and the Danes, and the largest (0.0667) is between the Lapps and the Sardinians. The mean genetic distance among the 861 available pairings of the 42 selected populations was found to be 0.1338..
The following table shows Fst calculated by Cavalli-Sforza (1994) for some populations:
| Bantu | Nio-Saharan | W. African | Mbuti | Japanese | Korean | Thai | Filipino | S. Chinese | Danish | English | Melanesian | New Guinean | Australian | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bantu | 0.00 | |||||||||||||
| Nio-Saharan | 0.01 | 0.00 | ||||||||||||
| W. African | 0.02 | 0.02 | 0.00 | |||||||||||
| Mbuti | 0.07 | 0.08 | 0.08 | 0.00 | ||||||||||
| Japanese | 0.24 | 0.25 | 0.23 | 0.31 | 0.00 | |||||||||
| Korean | 0.27 | 0.24 | 0.18 | 0.30 | 0.01 | 0.00 | ||||||||
| Thai | 0.34 | 0.30 | 0.25 | 0.39 | 0.07 | 0.06 | 0.00 | |||||||
| Filipino | 0.29 | 0.30 | 0.23 | 0.38 | 0.10 | 0.12 | 0.06 | 0.00 | ||||||
| S. Chinese | 0.30 | 0.29 | 0.20 | 0.34 | 0.05 | 0.05 | 0.01 | 0.03 | 0.00 | |||||
| Danish | 0.17 | 0.17 | 0.15 | 0.15 | 0.12 | 0.09 | 0.13 | 0.13 | 0.13 | 0.00 | ||||
| English | 0.23 | 0.18 | 0.15 | 0.24 | 0.12 | 0.10 | 0.11 | 0.11 | 0.12 | 0.00 | 0.00 | |||
| Melanesian | 0.34 | 0.31 | 0.27 | 0.40 | 0.11 | 0.11 | 0.08 | 0.05 | 0.06 | 0.14 | 0.16 | 0.00 | ||
| New Guinean | 0.34 | 0.33 | 0.28 | 0.46 | 0.12 | 0.14 | 0.18 | 0.13 | 0.15 | 0.16 | 0.16 | 0.07 | 0.00 | |
| Australian | 0.33 | 0.36 | 0.27 | 0.43 | 0.06 | 0.07 | 0.13 | 0.13 | 0.11 | 0.14 | 0.15 | 0.11 | 0.10 | 0.00 |
Autosomal genetic distances based on SNPs
A 2012 study based on International HapMap Project data estimated FST between the three major "continental" populations of Europeans (combined from Utah residents of Northern and Western European ancestry from the CEPH collection and Italians from Tuscany), East Asians (combining Han Chinese from Beijing, Chinese from metropolitan Denver and Japanese from Tokyo, Japan) and Sub-Saharan Africans (combining Luhya of Webuye, Kenya, Maasai of Kinyawa, Kenya and Yoruba of Ibadan, Nigeria). It reported a value close to 12% between continental populations, and values close to panmixia (smaller than 1%) within continental populations.[11]
| Europe (CEU) | Sub-Saharan Africa (Yoruba) | East-Asia (Japanese) | |
|---|---|---|---|
| Sub-Saharan Africa (Yoruba) | 0.153 | ||
| East-Asia (Japanese) | 0.111 | 0.190 | |
| East-Asia (Chinese) | 0.110 | 0.192 | 0.007 |
| Italians | Palestinians | Swedish | Finns | Spanish | Germans | Russians | French | Greeks | |
|---|---|---|---|---|---|---|---|---|---|
| Palestinians | 0.0064 | ||||||||
| Swedish | 0.0064-0.0090 | 0.0191 | |||||||
| Finns | 0.0130-0.0230 | 0.0050-0.0110 | |||||||
| Spanish | 0.0010-0.0050 | 0.0101 | 0.0040-0055 | 0.0110-0.0170 | |||||
| Germans | 0.0029-0.0080 | 0.0136 | 0.0007-0.0010 | 0.0060-0.0130 | 0.0015-0.0030 | ||||
| Russians | 0.0088-0.0120 | 0.0202 | 0.0030-0.0036 | 0.0060-0.0120 | 0.0070-0.0079 | 0.0030-0.0037 | |||
| French | 0.0030-0.0050 | 0.0020 | 0.0080-0.0150 | 0.0010 | 0.0010 | 0.0050 | |||
| Greeks | 0.0000 | 0.0057 | 0.0084 | 0.0035 | 0.0039 | 0.0108 | |||
Programs for calculating FST
Modules for calculating FST
References
- ↑ Peter Beerli, Estimation of migration rates and population sizes in geographically structured populations (1998), Advances in molecular ecology (ed. G. Carvalho). NATO Science Series A: Life Sciences, IOS Press, Amsterdam, 39-53.
- ↑ Frankham, R., Ballou, J.D., Briscoe, D.A., 2002. Introduction to Conservation Genetics. Cambridge University Press, Cambridge. Hartl DL, Clark AG (1997) Principles of Population Genetics, 3nd edn. Sinauer Associates, Inc, Sunderland, MA.
- ↑ B. M. von Holdt et al., "Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf", Science Advances 27 Jul 2016: Vol. 2, no. 7, e1501714, doi:10.1126/sciadv.1501714.
- ↑ Holsinger, Kent E.; Bruce S. Weir (2009). "Genetics in geographically structured populations: defining, estimating and interpreting FST". Nat Rev Genet. 10 (9): 639–650. doi:10.1038/nrg2611. ISSN 1471-0056. PMC 4687486. PMID 19687804.
- ↑ Richard Durrett (12 August 2008). Probability Models for DNA Sequence Evolution. Springer. ISBN 978-0-387-78168-6. Retrieved 25 October 2012.
- ↑ Hudson, RR.; Slatkin, M.; Maddison, WP. (Oct 1992). "Estimation of Levels of Gene Flow from DNA Sequence Data". Genetics. 132 (2): 583–9. doi:10.1093/genetics/132.2.583. PMC 1205159. PMID 1427045.
- ↑ Weir, B. S.; Cockerham, C. Clark (1984). "Estimating F-Statistics for the Analysis of Population Structure". Evolution. 38 (6): 1358–1370. doi:10.2307/2408641. ISSN 0014-3820. JSTOR 2408641. PMID 28563791.
- ↑ Cavalli-Sforza et al. (1994), cited after V. Ginsburgh, S. Weber, The Palgrave Handbook of Economics and Language, Springer (2016), p. 182.
- ↑ Harpending, Henry (2002). "Kinship and Population Subdivision". Population and Environment. 24 (2): 141–147. doi:10.1023/A:1020815420693. S2CID 15208802.
- ↑ Cavalli-Sforza et al., 1994, p. 24
- ↑ Elhaik, Eran (2012). "Empirical Distributions of FST from Large-Scale Human Polymorphism Data". PLOS ONE. 7 (11): e49837. Bibcode:2012PLoSO...749837E. doi:10.1371/journal.pone.0049837. PMC 3504095. PMID 23185452.
- 1 2 Nelis, Mari; et al. (2009-05-08). Fleischer, Robert C. (ed.). "Genetic Structure of Europeans: A View from the North–East". PLOS ONE. 4 (5): e5472. Bibcode:2009PLoSO...4.5472N. doi:10.1371/journal.pone.0005472. PMC 2675054. PMID 19424496., see table
- ↑ Tian, Chao; et al. (November 2009). "European Population Genetic Substructure: Further Definition of Ancestry Informative Markers for Distinguishing among Diverse European Ethnic Groups". Molecular Medicine. 15 (11–12): 371–383. doi:10.2119/molmed.2009.00094. ISSN 1076-1551. PMC 2730349. PMID 19707526., see table
- ↑ Crawford, Nicholas G. (2010). "smogd: software for the measurement of genetic diversity". Molecular Ecology Resources. 10 (3): 556–557. doi:10.1111/j.1755-0998.2009.02801.x. PMID 21565057. S2CID 205970662.
- ↑ Kitada S, Kitakado T, Kishino H (2007). "Empirical Bayes inference of pairwise F(ST) and its distribution in the genome". Genetics. 177 (2): 861–73. doi:10.1534/genetics.107.077263. PMC 2034649. PMID 17660541.
Further reading
- Evolution and the Genetics of Populations Volume 2: the Theory of Gene Frequencies, pg 294–295, S. Wright, Univ. of Chicago Press, Chicago, 1969
- A haplotype map of the human genome, The International HapMap Consortium, Nature 2005