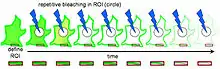
Fluorescence Loss in Photobleaching (FLIP) is a fluorescence microscopy technique used to examine movement of molecules inside cells and membranes. A cell membrane is typically labeled with a fluorescent dye to allow for observation. A specific area of this labeled section is then bleached several times using the beam of a confocal laser scanning microscope. After each imaging scan, bleaching occurs again. This occurs several times, to ensure that all accessible fluorophores are bleached since unbleached fluorophores are exchanged for bleached fluorophores, causing movement through the cell membrane. The amount of fluorescence from that region is then measured over a period of time to determine the results of the photobleaching on the cell as a whole.
Experimental Setup
Before photobleaching can occur, cells must be injected with a fluorescent protein, often a green fluorescent protein (GFP), which will allow the targeted proteins to fluoresce and therefore be followed throughout the process. Then, a region of interest must be defined. This initial region of interest usually contains the whole cell or several cells. In FLIP, photobleaching occurs just outside the region of interest; therefore a photobleaching region also needs to be defined. A third region, the region where measurement will take place, needs to be determined as well. A number of initial scans need to be made to determine fluorescence before photobleaching. These scans will serve as the control scans, to which the photobleached scans will be compared later on. Photobleaching can then occur. Between each bleach pulse, it is necessary to allow time for recovery of fluorescent material. It is also important to take several scans of the region of interest immediately after each bleach pulse for further study.[1] The change in fluorescence at the region of interest can then be quantified in one of three ways. The most common is to choose the location, size and number of the regions of interest based on visual inspection of the image sets. The two other, rather new but more reliable approaches are either by detecting areas of different probe mobility on an individual image basis or by physical modeling of fluorescence loss from moving bodies.[2]
Loss of fluorescence is defined by the mobile fraction, or the fraction of fluorophores capable of recovering into a photobleached area, of the fluorescently labeled protein. Incomplete loss of fluorescence indicates that there are fluorophores that do not move or travel to the bleached area. This allows for definition of the immobile fraction, or the fraction of fluorophores incapable of recovering into a photobleached area, of fluorescent-labeled proteins. Immobility indicates that there are proteins that may be in compartments closed off from the rest of the cell, preventing them from being affected by the repeated photobleaching.[3]
Applications
Verifying Continuity of Membranous Organelles
The primary use of FLIP is to determine the continuity of membranous organelles. This continuity or lack thereof is determined by observing the amount of fluorescence in the region of interest. If there is a complete loss of fluorescence, this indicates that the organelles are continuous. However, if there is incomplete loss of fluorescence, then there is not continuity between the organelles. Instead, these organelles are compartmentalized and therefore closed off to the transfer of any photobleached fluorophores.[3] Continuity of the Golgi apparatus, endoplasmic reticulum, and nucleus have been verified using FLIP.
Exchange Rate Between the Nucleus and Cytoplasm
Two of the other, less frequently employed uses of FLIP are to determine how proteins are shuttled from the Cytoplasm to the Nucleus and then determine the rate at which this shuttling occurs. To determine what portions are involved and when in the shuttling process they are involved, continuous scans are observed. The sooner a part of the cytoplasm is used in the shuttling process, the more rapidly it experiences complete loss of fluorescence.[4] The resulting image of this process should be a completely photobleached cytoplasm. If the cell also participates in nuclear export from the nucleus to the cytoplasm, photobleaching will also occur in the nucleus.
The exchange rate between the nucleus and cytoplasm can also be determined from this type of data. In these instances, the region of photobleaching is within the nucleus. If shuttling occurs at a rapid pace, the fluorescence levels within the nuclear compartments will decrease rapidly through the frames taken. However, if shuttling occurs slowly, the fluorescence levels will remain unaffected or decrease only slightly.[4]
FLIP vs. FRAP
FLIP is often used and is closely associated with Fluorescence recovery after photobleaching (FRAP). The major difference between these two microscopy techniques is that FRAP involves the study of a cell’s ability to recover after a single photobleaching event whereas FLIP involves the study of how the loss of fluorescence spreads throughout the cell after multiple photobleaching events. This difference in purpose also leads to a difference in what parts of the cell are observed. In FRAP, the area that is actually photobleached is the area of interest. Conversely, in FLIP, the region of interest is just outside the region that is being photobleached. Another important difference is that in FRAP, there is a single photobleaching event and a recovery period to observe how well fluorophores move back to the bleached site. However, in FLIP, multiple photobleaching events occur to prevent the return of unbleached fluorophores to the bleaching region. Like FLIP, FRAP is used in the study of continuity of membranous organelles. FLIP and FRAP are often used together to determine the mobility of GFP-tagged proteins.[3] FLIP can also be used to measure the molecular transfer between regions of a cell regardless of the rate of movement. This allows for a more comprehensive analysis of protein trafficking within a cell. This differs from FRAP which is primarily useful for determining mobility of proteins in regions local to the photobleaching only.[5]
Potential Complications
There are several complications that are involved with both FLIP and FRAP. Since both forms of microscopy examine living cells, there is always the possibility that the cells will move, causing false results. The best way to avoid this is to use an alignment algorithm, which will compensate for any movement and eliminate a large portion of the error due to movement. In these experiments, it is also key to have a control group in order to adjust the results and correct the recovery curve for the overall loss in fluorescence.[3] Another way to minimize error is to keep photobleaching contained to a single region or area. This limitation will serve as a control and limit fluorescence loss due to photo-damage as compared to fluorescence loss due to the photobleaching.[3]
See also
References
- ↑ Robert C. Dickson; Michael Dean Mendenhall (2004). Signal Transduction Protocols (2 ed.). Springer Science & Business Media. pp. 300–304. ISBN 978-1-59259-816-8.
- ↑ Wüstner, Daniel; Lukasz M Solanko; Frederik W Lund; Daniel Sage; Hans J Schroll; Michael A Lomholt (13 November 2012). "Quantitative fluorescence loss in photobleaching for analysis of protein transport and aggregation". BMC Bioinformatics. 13: 296. doi:10.1186/1471-2105-13-296. PMC 3557157. PMID 23148417.
- 1 2 3 4 5 Ishikawa-Ankerhold, Hellen C.; Ankerhold, Richard; Drummen, Gregor P. C. (2012). "Advanced Fluorescence Microscopy Techniques—FRAP, FLIP, FLAP, FRET and FLIM". Molecules. 17 (4): 4047–4132. doi:10.3390/molecules17044047. PMC 6268795. PMID 22469598.
- 1 2 Davies, R.G.; D.A. Jans; K.M. Wagstaff (2010). "Use of fluorescence photobleaching techniques to measure the kinetics of intracellular transport" (PDF). Microscopy: Science, Technology, Applications, and Education. Archived from the original (PDF) on 2014-12-26. Retrieved 2012-11-25.
- ↑ Kitamura, Akira; Kubota, Hiroshi (December 2010). "Amyloid oligomers: dynamics and toxicity in the cytosol and nucleus". FEBS Journal. 277 (6): 1369–1379. doi:10.1111/j.1742-4658.2010.07570.x. PMID 20148962.