Fluorination with aminosulfuranes is a chemical reaction that transforms oxidized organic compounds into organofluorine compounds. Aminosulfuranes selectively exchange hydroxyl groups for fluorine, but are also capable of converting carbonyl groups, halides, silyl ethers, and other functionality into organofluorides.[1]
Introduction
Prior to the introduction of diethylaminosulfur trifluoride (DAST) in 1970 for the replacement of hydroxyl groups with fluoride, sulfur tetrafluoride was the reagent most commonly used to accomplish this transformation. However, sulfur tetrafluoride only reacts with the most acidic hydroxyl groups (its substrate scope is limited), and is difficult to handle, toxic, and capable of generating hydrogen fluoride upon hydrolysis. Thus, aminosulfurane reagents such as diethylaminosulfur trifluoride have largely replaced SF4 as the reagents of choice for replacement of hydroxyl groups with fluoride.[2]
-
HOR FR
(1)
Aminosulfuranes are usually prepared by reaction of the corresponding dialkylamino(trialkyl)silanes with SF4.[2] When the aminosulfurane is exposed to a second equivalent of aminosilane, bis(dialkylamino)sulfur difluorides result.[3] Tris(dialkylamino)sulfonium difluorotrimethylsilicates such as tris(diethylamino)sulfonium difluorotrimethylsilicate (TASF) have achieved synthetic utility as reagents for the fluorination of halides. These form when three equivalents of aminosilane are exposed to sulfur tetrafluoride.[4]
-
(2)
![{\displaystyle {\begin{array}{l}{\ce {{R2N-Si(CH3)3}+ SF4 -> R2N-SF3}}\\{}\\{\ce {{R2N-SF3}->[{\ce {R2N-Si(CH3)3}}] R2N}}{-}\!{\begin{matrix}{\ce {F2}}\\{\ce {S}}\\{}\end{matrix}}\!{\ce {-NR2}}\\{\ce {{3R2N-Si(CH3)3}+ SF4 -> \underbrace{(R2N)2SF2+(CH3)3SiF2^-}_{TASF}}}\end{array}}}](../I/776821bd8121707d1ef3108ec4dedda8270846c7.svg)
Mechanism and stereochemistry
Prevailing mechanism
The mechanism of fluorination by DAST parallels that of sulfur tetrafluoride. Attack of the hydroxyl group of the substrate on sulfur and elimination of hydrogen fluoride lead to an alkoxyaminosulfur difluoride intermediate. Nucleophilic attack by fluoride, either by an SN1[5] or SN2[6] pathway, leads to the product. Although clean configurational inversion has been observed in a number of chiral alcohols, carbocationic rearrangements have also been observed in some cases. The operative pathway depends on the structure of the substrate.
-
(3)
![{\displaystyle {\begin{matrix}{}\\{\ce {R}}{-}{\color {Blue}{\ce {OH}}}\ {\ce {->[\color {Red}{\ce {R2N-SF3}}][-{\ce {H}}{\color {Red}{\ce {F}}}]R}}{-}{\color {Blue}{\ce {O}}}{\ce {-SF2NR2->[\color {Red}{\ce {F-}}][{\ce {S_{N}1\ or\ S_{N}2}}]R}}{-}{\color {Red}{\ce {F}}}\\{}\end{matrix}}}](../I/22904adda801c3a89197c72e6029a952bc3ba6dc.svg)
Conversion of aldehydes and ketones to the corresponding geminal difluorides proceeds by a similar mechanism, with addition of hydrogen fluoride preceding the hydroxyl replacement mechanism described above. An important side product in fluorinations of enolizable ketones is the corresponding vinyl fluoride, which results from deprotonation of intermediate fluoro carbocations.
-

(4)
Halides react by an essentially metathetical exchange of the halide for fluoride. Byproducts containing the exchanged halide have been isolated.[7]
Scope and limitations
Aminosulfuranes are highly selective for the replacement of hydroxyl groups with fluoride, but in the absence of alcohol functionality, they have the ability to transform a wide array of substrates into the corresponding fluorides or acyl fluorides. For example, ketones are converted to geminal difluorides.[8] However, unlike sulfur tetrafluoride, aminosulfuranes do not convert carboxylic acids into trifluoromethyl groups; the reaction halts at the acyl fluoride stage.[9] Silyl ethers are converted to organofluorides in the presence of DAST.[10]
-
(5)

Aldehydes and ketones react with DAST to form the corresponding geminal difluorides. Fluorination of enolizable ketones gives a mixture of the difluoroalkane and vinyl fluoride. In glyme with fuming sulfuric acid, the vinyl fluoride product predominates.[11] Electron-rich carbonyl compounds, such as esters and amides, do not react with DAST or other aminosulfuranes.
-
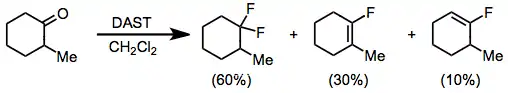
(6)
Epoxides may yield a variety of products depending on their structure. Generally, the products that form in highest yield are vicinal difluorides and bis(α-fluoroalkyl)ethers. However, this reaction results in low yields and is not synthetically useful.[12]
-
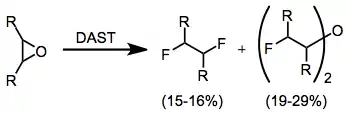
(7)
The polar mechanism of fluorination by DAST implies that certain substrates may suffer Wagner-Meerwein rearrangements. This process has been observed in the fluorination of pivalaldehyde, which affords a mixture of 1,2-difluoro-1,2-dimethylpropane, 1,1-difluoro-2,2-dimethylpropane, and 1-fluoro-2,2-dimethylethylene.[8]
-
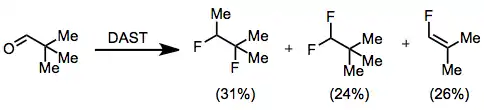
(8)
Diols can undergo pinacol rearrangement under fluorination conditions.[13]
-
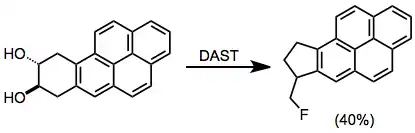
(9)
When sulfoxides are treated with DAST, an interesting Pummerer-type rearrangement occurs to afford α-fluoro sulfides.[14]
-
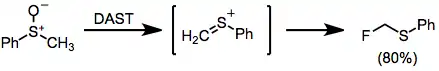
(10)
Comparison with other methods
Aminosulfuranes compare favorably with many of the other fluorination methods available. They are easier to handle than sulfur tetrafluoride; however SF4 does not promote cationic rearrangements.[15] With respect to carboxylic acids, aminosulfuranes and SF4 are complementary: the former gives acid fluorides, while the latter gives trifluoromethyl compounds.
-
(11)
![{\displaystyle {\ce {{\underset {carboxylic\ acid}{R-CO2H}}->[{} \atop {\ce {SF4}}]{\underset {trifluoromethyl}{R-CF3}}}}}](../I/fab5bbd5519e38953de667bb9c011406cfa65e14.svg)
Perfluorinated alkylamines, such as Ishikawa's reagent (N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine WRONG MOLECULE IN SCHEME BELOW),[16] are highly selective for hydroxyl groups and do not react with aldehydes and ketones. The amide byproducts of these reagents, however, are harder to separate from the desired products than aminosulfurane byproducts.
-
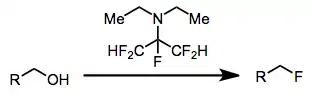
(12)
Alkali and tetraalkylammonium fluorides can be used to displace sulfonate esters; however, these reactions require higher temperatures than aminosulfurane fluorination of the corresponding free alcohols.[17]
-
ROSO
2R RF(13)
Experimental conditions and procedure
Typical conditions
Fluorinations with DAST can be carried out in conventional glass equipment, although etching of the glass may result from reaction byproducts. Reactions are typically carried out in aprotic or non-polar solvents. Moisture and atmospheric oxygen should be excluded from the reaction. Reactions are generally started at -78 °C and warmed to room temperature or above; however, reactions should not be heated above 80 °C, as decomposition of the fluorinating reagent begins to occur at this temperature. Workup usually involves pouring the reaction mixture over water or ice, followed by neutralization of acidic byproducts with sodium bicarbonate. Standard purification methods can be used to isolate the desired fluorinated products.
Example procedure
-
HOC
7H
15 FC
7H
15 (90%)(14)
A solution of 13.0 g (0.1 mol) of 1-octanol in 25 mL of dichloromethane was added dropwise to a solution of 16.1 g (0.1 mol) of diethylaminosulfur trifluoride in 60 mL of dichloromethane cooled to –70° to –65°. The reaction mixture was warmed to 25°, 50 mL of water was added, and the lower organic layer was separated and dried with anhydrous magnesium sulfate and distilled to give 12.0 g (90%) of 1-fluorooctane as a colorless liquid, bp 42–43° (20 mm). 19F NMR (CCl3F): -218.8 ppm (tt, 2J = 49 Hz, 3J = 25 Hz).[18]
References
- ↑ Hudlicky, M. Org. React. 1988, 35, 513. doi:10.1002/0471264180.or035.03
- 1 2 von Halasz, P.; Glemser, O. Chem. Ber. 1970, 103, 594.
- ↑ Markovskii, N.; Pashinnik, E.; Kirsanova, A. Zh. Org. Khim. 1975, 11, 74 (Engl. transl., 72).
- ↑ Middleton, W. J. U.S. Patent 3940402 (1976) [C.A., 85, 6388j (1976)].
- ↑ Rozen, S. ; Faust, Y. ; Ben-Yakov, H. Tetrahedron Lett. 1979, 20, 1823.
- ↑ Leroy, J.; Hebert, E.; Wakselman, C. J. Org. Chem. 1979, 44, 3406.
- ↑ Markovskii, N.; Pashinnik, E. Synthesis, 1975, 801.
- 1 2 Middleton, J. J. Org. Chem. 1975, 40, 574.
- ↑ Markovskii, N.; Pashinnik, E.; Kirsanova, A. Synthesis, 1973, 787.
- ↑ LeTourneau, E.; McCarthy, R. Tetrahedron Lett. 1984, 25, 5227.
- ↑ Boswell, Jr., G. A. U.S. Patent 4212815 (1980) [C.A., 93, 239789w (1980)].
- ↑ Hudlický, M. J. Fluorine Chem. 1987, 36, 373.
- ↑ Newman, S.; Khanna, M.; Kanakarajan, K. J. Am. Chem. Soc. 1979, 101, 6788.
- ↑ McCarthy, R.; Peet, P.; LeTourneau, E.; Inbasekaran, M. J. Am. Chem. Soc., 107, 735 (1985).
- ↑ Wang, C.-L. Org. React. 1985, 34, 319.
- ↑ Takaoka, A.; Iwagiri, H.; Ishikawa, N. Bull. Chem. Soc. Jpn. 1979, 52, 3377.
- ↑ Pattison, M.; Millington, E. Can. J. Chem. 1956, 34, 757.
- ↑ Middleton, W. J. U.S. Patent 3914265 (1975) [C.A., 84, 42635a (1976)]; 3976691 (1976) [C.A., 86, 29054g (1977)].