Genetic saturation is the result of multiple substitutions at the same site in a sequence, or identical substitutions in different sequences, such that the apparent sequence divergence rate is lower than the actual divergence that has occurred.[1] When comparing two or more genetic sequences consisting of single nucleotides, differences in sequence observed are only differences in the final state of the nucleotide sequence. Single nucleotides that undergoing genetic saturation change multiple times, sometimes back to their original nucleotide or to a nucleotide common to the compared genetic sequence. Without genetic information from intermediate taxa, it is difficult to know how much, or if any saturation has occurred on an observed sequence.[2] Genetic saturation occurs most rapidly on fast-evolving sequences, such as the hypervariable region of mitochondrial DNA, or in short tandem repeats such as on the Y-chromosome.[3][4]
In phylogenetics, saturation effects result in long branch attraction, where the most distant lineages have misleadingly short branch lengths. It also decreases phylogenetic information contained in the sequences.[5]
Phylogenetic saturation
Multiple substitutions
Multiple substitutions take place when single nucleotides undergo multiple changes before reaching their final nucleotide identity. A sequence is said to be saturated because mutation has acted multiple times upon nucleotides and observed change in sequence is, in fact, less than the historical change in sequence.[1]
Detection
It is possible to estimate the amount of saturation that a sequence might have undergone by estimating the substitution rate of a genetic sequence and how much time has passed since divergence. Divergence rates are estimated from a variety of sources including ancestral DNA, fossil records and biographical events.[6] This use of molecular clocks to determine divergence is controversial because of its potential for inaccuracy and assumptions made in the model (such as consistent mutation rate for all branches) and is used mostly as an estimation tool.[6] Genetic saturation can also be estimated by comparing the number of observed differences in nucleotide sequences between multiple pairs of species. The number of observed substitutions between sequences of different species can be compared to the number of inferred substitutions based on branch length to find the approximate point where the number of inferred substitutions surpasses the number of observed substitutions.[6][7] This method can give researchers an idea of the level of saturation of a particular gene but is thought to underestimate the amount of saturation, especially for very large branch lengths.[2]
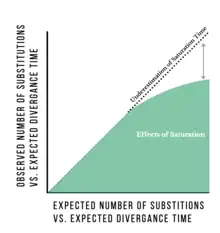
Impact on phylogenetics
In the field of molecular phylogenetics, the distances and relationships between species are investigated by looking at the DNA, RNA or amino acid sequences of an organism. When phylogenetic trees are constructed without considering possible saturation, the possibility of multiple substitutions can cause the distance between taxa to appear much smaller than the true distance. Multiple sequence alignment, a common technique to construct phylogenies, relies on the comparison of homologous sequences. It can easily be confounded by genetic saturation because the homologous loci under investigation show no indication whether or not more than one substitution on each nucleotide separates the taxa being described.[1] Substitution decreases the amount of phylogenetic information that can be contained in sequences, especially when deep branches are involved. This is particularly evident in studies examining arthropod groups.[8] Furthermore, saturation effects can lead to a gross underestimation of divergence time. This is mainly attributed to the randomization of the phylogenetic signal with the number of observed sequence mutations and substitutions. The effects of saturation can mask the true amount of divergence time leading to inaccurate phylogenetic trees.[1][2]
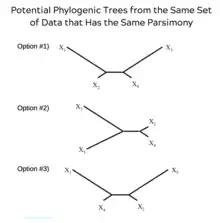
The principle of parsimony in genetic saturation analysis
Parsimony plays a fundamental role in genetic saturation analysis. This principle gives preference to the simplest explanation that can explain the data. In regards to genetic saturation, parsimony means that the hypothesized relationship is one that has the smallest number of character changes. Using parsimony to analyze genetic saturation can lead to conflict when creating a phylogenetic tree.[7] When only sequence data is used, it is possible to come up with numerous phylogenetic trees with the same amount of parsimony.
Long branch attraction
Genetic saturation contributes to long-branch attraction in its ability to greatly mix up genetic code without easily observable associated phenotypic changes. Long branch attraction occurs when two relatively outgrouped taxa are seemingly closely linked.[1] The more substitution mutations, the more likely it is for previously dissimilar sequences to share nucleotides and as a result, show homology in phylogenetic tree calculations. Long-branch attraction due to saturation has been proposed to be the cause of links in ancient phylogenies and puts into question even some of the earliest relationships between eukaryotes, archaea, and eubacteria. [2]
Other uses of "Saturation" in genetics
Gene site saturation mutagenesis
Gene site saturation mutagenesis (GSSM) is mutagenesis technique of one or more codons in a gene to create a library of variants covering all other codons at that position.[9] It is used in biochemistry and protein engineering to explore the functions and characteristics of specific amino acid sequences.[9] This systemic identification of amino acid substitutions allows researchers to look at every possible variant of each position. This will provide crucial structural information about the protein of interest and will identify amino acid sequences that are more vital to the function of the protein.[9][10]

Researchers often lean towards using a one-step PCR-based to explore the specific effects of different variations in an amino acid of interest within a protein with GSSM.[11] With a one-step PCR-based approached, researchers create a primer that has a corresponding sequence to the protein of interest at its two ends. Only one codon of a three codon amino acid sequence is substituted.[10]
The type of codon set, will determine the number of sequences that can be derived from GSSM. To determine which codon set to use, researchers will need to check the library quality on the DNA level, which means that massive sequence data is needed. If all 3 positions can be substituted for each of the four different nucleotides, researchers can code for all 20 amino acids.[10] Although it’s possible to code for all 20 amino acids, this is not the most efficient method. The most efficient method is to use an NNK codon degeneracy, also known as a limited codon set.[12] This method, will result in only 32 codons rather than 64.[10]
Advantages of GSSM
In comparison to other techniques, GSSM is able to offer unique advantages such as:
- A complete analysis of every position in a given gene, which can be helpful in identifying critical positions. Critical positions are identified by analyzing the immensity of the effects of mutagenesis — both positive and negative. GSSM can also identify positions that are more flexible, as GSSM at these positions will have less of an impact on the amino acid.[9]
- A residue-specific analysis, which allows for researchers to create a schematic representation of the amino acid. This allows for more complex and detailed genetic research in further studies.[9]
- An ability to look at the effects of various amino acids without knowing any structural information about the protein. The data collected can then provide valuable insight into this area. [9]
- Fast delivery times and cost-efficiency.[9]
GSSM was able to open up a whole frontier in genetic research, as it revolutionized fundamental beliefs about DNA. Before GSSM, researchers mutated DNA through radiation or with various chemicals. Both of these methods are imprecise.[13]
References
- 1 2 3 4 5 Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Wörheide G, Baurain D (March 2011). "Resolving difficult phylogenetic questions: why more sequences are not enough". PLOS Biology. 9 (3): e1000602. doi:10.1371/journal.pbio.1000602. PMC 3057953. PMID 21423652.
- 1 2 3 4 Philippe H, Forterre P (October 1999). "The rooting of the universal tree of life is not reliable". Journal of Molecular Evolution. 49 (4): 509–23. Bibcode:1999JMolE..49..509P. doi:10.1007/PL00006573. PMID 10486008. S2CID 20350374.
- ↑ Henn BM, Gignoux CR, Feldman MW, Mountain JL (January 2009). "Characterizing the time dependency of human mitochondrial DNA mutation rate estimates". Molecular Biology and Evolution. 26 (1): 217–30. doi:10.1093/molbev/msn244. PMID 18984905.
- ↑ Ho SY, Phillips MJ, Cooper A, Drummond AJ (July 2005). "Time dependency of molecular rate estimates and systematic overestimation of recent divergence times". Molecular Biology and Evolution. 22 (7): 1561–8. doi:10.1093/molbev/msi145. PMID 15814826.
- ↑ Abylgazieva NA (2003-01-01). "[Case of "renal diabetes"]". Zdravookhranenie Kirgizii. 26 (3): 49–51. doi:10.1016/S1055-7903(02)00326-3. PMID 7903.
- 1 2 3 van Tuinen M, Dyke GJ (January 2004). "Calibration of galliform molecular clocks using multiple fossils and genetic partitions". Molecular Phylogenetics and Evolution. 30 (1): 74–86. doi:10.1016/S1055-7903(03)00164-7. PMID 15022759.
- 1 2 Dávalos LM, Perkins SL (May 2008). "Saturation and base composition bias explain phylogenomic conflict in Plasmodium". Genomics. 91 (5): 433–42. doi:10.1016/j.ygeno.2008.01.006. PMID 18313259.
- ↑ Sanders KL, Lee MS (April 20, 2009). "Arthropod molecular divergence times and the Cambrian origin of pentastomids". Systematics and Biodiversity. 8 (1): 63–74. doi:10.1080/14772000903562012. S2CID 84880682.
- 1 2 3 4 5 6 7 Zheng L, Baumann U, Reymond JL (August 2004). "An efficient one-step site-directed and site-saturation mutagenesis protocol". Nucleic Acids Research. 32 (14): e115. doi:10.1093/nar/gnh110. PMC 514394. PMID 15304544.
- 1 2 3 4 Lopez P, Forterre P, Philippe H (October 1999). "The root of the tree of life in the light of the covarion model". Journal of Molecular Evolution. 49 (4): 496–508. Bibcode:1999JMolE..49..496L. doi:10.1007/pl00006572. PMID 10486007. S2CID 22835829.
- ↑ Li A, Acevedo-Rocha CG, Reetz MT (July 2018). "Boosting the efficiency of site-saturation mutagenesis for a difficult-to-randomize gene by a two-step PCR strategy". Applied Microbiology and Biotechnology. 102 (14): 6095–6103. doi:10.1007/s00253-018-9041-2. PMC 6013526. PMID 29785500.
- ↑ Kretz KA, Richardson TH, Gray KA, Robertson DE, Tan X, Short JM (Aug 6, 2004). "Gene site saturation mutagenesis: a comprehensive mutagenesis approach". Protein Engineering. Methods in Enzymology. Vol. 388. pp. 3–11. doi:10.1016/S0076-6879(04)88001-7. ISBN 9780121827939. PMID 15289056.
- ↑ Smith I, Payne J, Keay B. "How Michael Smith put B.C.'s life sciences community on the map with a Nobel Prize 25 years ago". Vancouver Sun. Retrieved 24 September 2018.