Glyceroneogenesis is a metabolic pathway which synthesizes glycerol 3-phosphate (used to form triglycerides) from precursors other than glucose.[1] Usually, glycerol 3-phosphate is generated from glucose by glycolysis, in the liquid of the cell's cytoplasm (the cytosol). Glyceroneogenesis is used when the concentrations of glucose in the cytosol are low, and typically uses pyruvate as the precursor, but can also use alanine, glutamine, or any substances from the TCA cycle. The main regulator enzyme for this pathway is an enzyme called phosphoenolpyruvate carboxykinase (PEPC-K), which catalyzes the decarboxylation of oxaloacetate to phosphoenolpyruvate.[1] Glyceroneogenesis is observed mainly in adipose tissue, and in the liver. A significant biochemical pathway regulates cytosolic lipid levels. Intense suppression of glyceroneogenesis may lead to metabolic disorders such as type 2 diabetes.[2]
Summary
Triglycerides are built from three fatty acids, esterified onto each of three hydroxy groups of glycerol, which is derived from glycerol 3-phosphate. In mammals, glycerol 3-phosphate is usually synthesized through glycolysis, a metabolic pathway that degrades glucose into fructose 1,6-bisphosphate and then into two molecules of dihydroxyacetone phosphate, which beget glycerol 3-phosphate and glyceraldehyde 3-phosphate.[1] When an organism is deficient in glucose, from (for example) fasting or a low carbohydrate intake, glycerol 3-phosphate is generated by glyceroneogenesis instead. As well as synthesizing lipids for use in other metabolic processes, glyceroneogenesis regulates lipid levels in the cytosol.[1]
Metabolic pathway
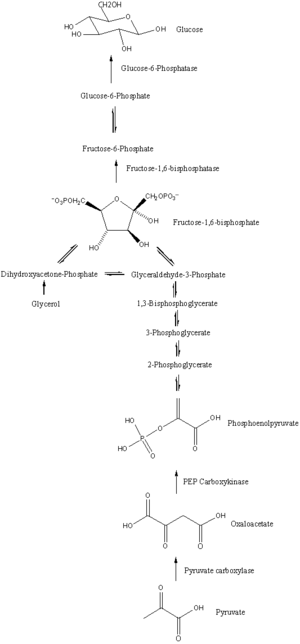
The main precursors of glyceroneogenesis are pyruvate, lactate, glutamine, and alanine. Glyceroneogenesis is also known as the branched pathway of gluconeogenesis because its first few steps are the same.
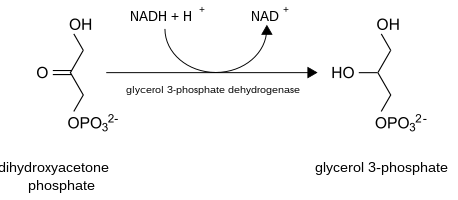
When pyruvate or lactate is used as the precursor for glycerol 3-phosphate, glyceroneogenesis follows the same pathway as gluconeogenesis until it generates dihydroxyacetone phosphate. Lactate catalyzed by lactate dehydrogenase will form pyruvate at the expense of NAD+. By using one ATP and bicarbonate, pyruvate will be converted to oxaloacetate, catalysed by pyruvate carboxylase. The PEPC-K enzyme will catalyze oxaloacetate to generate phosphoenolpyruvate. This phosphorylation and decarboxylation of oxaloacetate is a significant step in glyceroneogenesis, since it regulates the entire pathway. After the production of phosphoenolpyruvate, gluconeogenesis will continue until dihydroxyacetone phosphate is generated, which produces 2-phosphoglycerate, 3-phosphoglycerate, 1,3-bisphosphoglycerate and glyceraldehyde 3-phosphate as intermediates. When dihydroxyacetone phosphate is produced, glyceroneogenesis will branch off from gluconeogenesis.[1] With the expense of NADH, dihydroxyacetone phosphate will convert to glycerol 3-phosphate, which is the final product of glyceroneogenesis. In addition, triglyceride can be generated by re-esterifying 3 fatty acid chains on glycerol 3-phosphate. Instead of producing fructose 1,6- bisphosphate as gluconeogenesis does, glyceroneogenesis converts dihydroxyacetone phosphate to glycerol 3-phosphate.
Alanine can also be used as a precursor of glyceroneogenesis because alanine can be degraded to pyruvate. Alanine will degrade to pyruvate by transferring its amino group to 2-oxoglutarate with an enzyme called alanine aminotransferase. Alanine aminotransferase cleaves off the amino group from alanine and binds it to 2-oxoglutarate, generating pyruvate from alanine, and glutamate from 2-oxoglutarate. Pyruvate generated from alanine will enter glyceroneogenesis and generate glycerol 3-phosphate.
Glutamate can also enter glyceroneogenesis. Since the key reaction of glyceroneogenesis is the decarboxylation and phosphorylation of oxaloacetate to phosphoenolpyruvate, in theory any biochemical pathway which generates oxaloacetate is related to glyceroneogenesis. For example, glutamate can generate oxaloacetate in 2 steps. Firstly, glutamate can be converted to 2-oxoglutarate with the expense of NAD+ and H2O with the help of glutamate dehydrogenase. Secondly, 2-oxoglutarate can enter the tricarboxylic acid cycle to generate oxaloacetate. Therefore, theoretically any metabolites in the TCA cycle or any metabolites generating the metabolites of the TCA cycle can be used as a precursor of glyceroneogenesis, but glutamate is the only precursor confirmed.
Regulation
Phosphoenolpyruvate carboxykinase (PEPC-K)
Glyceroneogenesis can be regulated at two reaction pathways. First, it can be held at the decarboxylation of oxaloacetate to phosphoenolpyruvate. Secondly, the TCA cycle can affect glyceroneogenesis when the glutamate or substrates in the TCA cycle are being used as a precursor. Decarboxylation of oxaloacetate to phosphoenolpyruvate is catalyzed by PEPC-K, the essential enzyme which regulates glyceroneogenesis.[1] Increases in PEPC-K levels or overexpression of the gene that codes for PEPC-K will increase glyceroneogenesis. Also, oxaloacetate can be decarboxylated to phosphoenolpyruvate when more PEPC-K can catalyze the reaction.
Gene expression of PEPC-K can be suppressed by norepinephrine, glucocorticoids, and insulin.[3] Norepinephrine is a neurotransmitter which decreases the activity of PEPC-K when the cell is in a cold environment. Glucocorticoids are steroid hormones involved in the reciprocal regulation of glyceroneogenesis in the liver and adipose tissues. Through a poorly-understood mechanism, they induce transcription of PEPC-K in the liver while decreasing transcription in adipose tissues. Insulin is a peptide hormone that causes cells to take in glucose. Through glyceroneogenesis, insulin down-regulates the expression of PEPC-K in both liver and adipose tissues.
TCA cycle
When metabolites from the TCA cycle or glutamate are used as a precursor for glyceroneogenesis, the regulator in the TCA cycle can also cause fluctuations in the levels of products formed by glyceroneogenesis. Regulation of the TCA cycle is mainly determined by product inhibition and substrate availability. The TCA cycle will slow down when the environment contains excess product, or deficiency of the substrate such as ADP and NAD+.
Location
Since glyceroneogenesis is related to lipid regulation, it can be found in adipose tissue and the liver. In adipose tissue, glyceroneogenesis restrains the release of free fatty acids (FFA) by re-esterifying them. In the liver, triglycerides are synthesized for lipid distribution.
White adipose tissue
White adipose tissue, also known as white fat, is one two types of adipose tissue in mammals. White adipose tissue stores energy in the form of triglycerides, which can be broken down to free fatty acids on demand. Its normal function is to store free fatty acids as triglycerides within the tissue. When glucose is deficient, in situations like fasting, white adipose tissue generates glycerol 3-phosphate.[3]
Brown adipose tissue
Brown adipose tissue stores free fatty acids rather than triglycerides, and is especially abundant in newborn and hibernating mammals. Brown adipose tissue is involved in thermogenesis, and has a considerably higher glyceroneogenesis activity.[3] Brown adipose tissue contains more glyceroneogenesis-related enzymes, in particular PEPC-K and glycerol kinase. PEPC-K is around 10 times more active than in white adipose tissue, and is the key regulatory enzyme that controls the activity of the pathway.[3] Glycerol kinase phosphorylates glycerol to generate glycerol 3-phosphate, which is used to build triglycerides. An increase in the activity of glycerol kinase will increase the production of glycerol 3-phosphate.
Glyceroneogenesis in brown adipose tissue contributes to thermogenesis, a process that generates heat in warm-blooded animals by delivering free fatty acids to the mitochondria.[3] In normal conditions, thermogenesis is down-regulated by the low concentration of free fatty acids in the cytosol, because glyceroneogenesis re-esterifies fatty acids to triglycerides. When exposed to cold, a neurotransmitter hormone called norepinephrine suppresses the activity of PEPC-K and thus the glyceroneogenesis re-esterification, increasing the availability of free fatty acids within the cell.[3] Excess free fatty acids in the cytosol will consequently be delivered to the mitochondria for thermogenesis.[4]
Liver
Although glyceroneogenesis was first found in adipose tissues, it was not recognized in the liver until 1998. This finding was unexpected because triglyceride synthesis in the liver was thought not to occur due to the amount of gluconeogenesis taking place, and because the liver was believed to have sufficient glycerol 3-phosphate collected from the bloodstream. Several experiments using stable isotopes to track the glycerol in the liver and bloodstream, showed that 65% of the glycerol backbone of triglycerides in the bloodstream is synthesized in the liver.[3] It was subsequently found that the liver synthesizes more than half of the glycerol mammals need to regulate lipids.
Glyceroneogenesis in the liver and adipose tissues regulate lipid metabolism in opposite ways. Lipids as triglycerides are released from the liver, while glyceroneogenesis restrains the fatty acid release from adipose tissues by re-esterifying them.[3] When the lipid concentration in the blood is relatively high, glyceroneogenesis in the liver will be down-regulated to stop the synthesis of triglycerides, but glyceroneogenesis in adipose tissues will be induced in order to restrain the release of free fatty acid to the bloodstream. Conversely, glyceroneogenesis is induced in the liver and suppressed in adipose tissues when the blood lipid level is low. Although the reciprocal regulation of glyceroneogenesis is not well understood, a hormone called glucocorticoid is involved in the regulation.[4] Glucocorticoids induce gene transcription of PEPC-K in liver but repress the transcription in adipose tissues.
Disease
Type 2 Diabetes
Failure in the regulation of glyceroneogenesis may lead to type 2 diabetes, a metabolic disorder that results in high levels of blood glucose and blood lipid.[5] Type 2 diabetes, in addition to a decreased sensitivity to insulin, is associated with the overproduction of triglycerides in the liver, due to excessively active glyceroneogenesis and excess release of fatty acids from adipose tissues. Glyceroneogenesis can be regulated by controlling the gene expression of PEPC-K.
Overexpressing PEPC-K in the liver will overproduce triglycerides and elevate the lipid level in the bloodstream, increasing the risk of fatty liver disease (hepatic steatosis). Conversely, in adipose tissue, down-regulated glyceroneogenesis may decrease de novo lipogenesis, increasing the export of free fatty acids to the bloodstream, leading to lipodystrophy. Both of these conditions are highly associated with type 2 diabetes.
Treatment
Regulation of glyceroneogenesis is a therapeutic target of type 2 diabetes treatment, specifically inhibiting it in the liver and increasing it in adipose tissues. Insulin down-regulates glyceroneogenesis in the liver, but it also suppresses it in adipose tissue. To restrict the release of free fatty acids from adipose tissues, glyceroneogenesis must be increased so they are re-esterified. Thiazolidinedione is a substance that only affects glyceroneogenesis in adipose tissue by increasing transcription of PEPC-K to up-regulate glyceroneogenesis.[5]
See also
References
- 1 2 3 4 5 6 Nye CK, Hanson RW, Kalhan SC (October 2008). "Glyceroneogenesis is the dominant pathway for triglyceride glycerol synthesis in vivo in the rat". The Journal of Biological Chemistry. 283 (41): 27565–74. doi:10.1074/jbc.M804393200. PMC 2562054. PMID 18662986.
- ↑ Jeoung NH, Harris RA (October 2010). "Role of pyruvate dehydrogenase kinase 4 in the regulation of blood glucose levels". Korean Diabetes Journal. 34 (5): 274–83. doi:10.4093/kdj.2010.34.5.274. PMC 2972486. PMID 21076574.
- 1 2 3 4 5 6 7 8 Reshef L, Olswang Y, Cassuto H, Blum B, Croniger CM, Kalhan SC, Tilghman SM, Hanson RW (August 2003). "Glyceroneogenesis and the triglyceride/fatty acid cycle". The Journal of Biological Chemistry. 278 (33): 30413–6. doi:10.1074/jbc.R300017200. PMID 12788931.
- 1 2 Chaves VE, Frasson D, Martins-Santos ME, Boschini RP, Garófalo MA, Festuccia WT, Kettelhut IC, Migliorini RH (October 2006). "Glyceroneogenesis is reduced and glucose uptake is increased in adipose tissue from cafeteria diet-fed rats independently of tissue sympathetic innervation". The Journal of Nutrition. 136 (10): 2475–80. doi:10.1093/jn/136.10.2475. PMID 16988112.
- 1 2 Beale EG, Hammer RE, Antoine B, Forest C (April 2004). "Disregulated glyceroneogenesis: PCK1 as candidate diabetes and obesity gene". Trends in Endocrinology and Metabolism. 15 (3): 129–35. doi:10.1016/j.tem.2004.02.006. PMID 15046742. S2CID 9194909.
Metabolism map | ||
|---|---|---|
Single lines: pathways common to most lifeforms. Double lines: pathways not in humans (occurs in e.g. plants, fungi, prokaryotes). | ||
.svg.png.webp)