The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a simple method for calculating molecular orbitals as linear combinations of atomic orbitals. The theory predicts the molecular orbitals for π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine.[1][2][3] It provides the theoretical basis for Hückel's rule that cyclic, planar molecules or ions with π-electrons are aromatic. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon and hydrogen (heteroatoms).[4] A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general (not just planar systems) and was used to provide computational justification for the Woodward–Hoffmann rules.[5] To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM).

Although undeniably a cornerstone of organic chemistry, Hückel's concepts were undeservedly unrecognized for two decades. Pauling and Wheland characterized his approach as "cumbersome" at the time, and their competing resonance theory was relatively easier to understand for chemists without fundamental physics background, even if they couldn't grasp the concept of quantum superposition and confused it with tautomerism. His lack of communication skills contributed: when Robert Robinson sent him a friendly request, he responded arrogantly that he is not interested in organic chemistry.[6]
In spite of its simplicity, the Hückel method in its original form makes qualitatively accurate and chemically useful predictions for many common molecules and is therefore a powerful and widely taught educational tool. It is described in many introductory quantum chemistry and physical organic chemistry textbooks, and organic chemists in particular still routinely apply Hückel theory to obtain a very approximate, back-of-the-envelope understanding of π-bonding.
Hückel characteristics
The method has several characteristics:
- It limits itself to conjugated molecules.
- Only π electron molecular orbitals are included because these determine much of the chemical and spectral properties of these molecules. The σ electrons are assumed to form the framework of the molecule and σ connectivity is used to determine whether two π orbitals interact. However, the orbitals formed by σ electrons are ignored and assumed not to interact with π electrons. This is referred to as σ-π separability. It is justified by the orthogonality of σ and π orbitals in planar molecules. For this reason, the Hückel method is limited to systems that are planar or nearly so.
- The method is based on applying the variational method to linear combination of atomic orbitals and making simplifying assumptions regarding the overlap, resonance and Coulomb integrals of these atomic orbitals. It does not attempt to solve the Schrödinger equation, and neither the functional form of the basis atomic orbitals nor details of the Hamiltonian are involved.
- For hydrocarbons, the method takes atomic connectivity as the only input; empirical parameters are only needed when heteroatoms are introduced.
- The method predicts how many energy levels exist for a given molecule, which levels are degenerate and it expresses the molecular orbital energies in terms of two parameters, called α, the energy of an electron in a 2p orbital, and β, the interaction energy between two 2p orbitals (the extent to which an electron is stabilized by allowing it to delocalize between two orbitals). The usual sign convention is to let both α and β be negative numbers. To understand and compare systems in a qualitative or even semi-quantitative sense, explicit numerical values for these parameters are typically not required.
- In addition the method also enables calculation of charge density for each atom in the π framework, the fractional bond order between any two atoms, and the overall molecular dipole moment.
Hückel results
Results for simple molecules and general results for cyclic and linear systems
The results for a few simple molecules are tabulated below:
| Molecule | Energy | Frontier orbital | HOMO–LUMO energy gap | Notes |
|---|---|---|---|---|
| E1 = α + β | HOMO | 2β | ||
| E2 = α – β | LUMO | |||
| E1 = α + 1.618...β | 1.236...β | 1.618... and 0.618... = | ||
| E2 = α + 0.618...β | HOMO | |||
| E3 = α – 0.618...β | LUMO | |||
| E4 = α – 1.618...β | ||||
| E1 = α + 1.802...β | 0.890...β | 1.802..., 1.247..., and 0.445... = 2cos(nπ/7) for n = 1, 2, and 3 | ||
| E2 = α + 1.247...β | ||||
| E3 = α + 0.445...β | HOMO | |||
| E4 = α – 0.445...β | LUMO | |||
| E5 = α – 1.247...β | ||||
| E6 = α – 1.802...β | ||||
| E1 = α + 2β | 0 | (E2, E3) are degenerate, both are singly occupied for D4h (square) cyclobutadiene, to comply with Hund's rule[7] | ||
| E2 = α | SOMO | |||
| E3 = α | SOMO | |||
| E4 = α − 2β | ||||
| E1 = α + 2β | 2β | (E2, E3) and (E4, E5) are degenerate | ||
| E2 = α + β | HOMO | |||
| E3 = α + β | HOMO | |||
| E4 = α − β | LUMO | |||
| E5 = α − β | LUMO | |||
| E6 = α − 2β | ||||
| Table 1. Hückel method results. Because α and β are negative,[8] orbitals are in order of increasing energy.
HOMO/LUMO/SOMO = Highest occupied/lowest unoccupied/singly-occupied molecular orbitals. |
||||

The theory predicts two energy levels for ethylene with its two π electrons filling the low-energy HOMO and the high energy LUMO remaining empty. In butadiene the 4 π-electrons occupy 2 low energy molecular orbitals, out of a total of 4, and for benzene 6 energy levels are predicted, two of them degenerate.
For linear and cyclic systems (with N atoms), general solutions exist:[9]
- Linear system (polyene/polyenyl): .
- Energy levels are all distinct.
- Linear system (polyene/polyenyl): .

- Cyclic system, Hückel topology (annulene/annulenyl): .
- Energy levels are each doubly degenerate.
- Cyclic system, Möbius topology (hypothetical for N < 8[10]): .
- Energy levels are each doubly degenerate.
- Cyclic system, Hückel topology (annulene/annulenyl): .




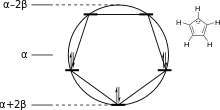
The energy levels for cyclic systems can be predicted using the Frost circle mnemonic (named after the American chemist Arthur Atwater Frost). A circle centered at α with radius 2β is inscribed with a regular N-gon with one vertex pointing down; the y-coordinate of the vertices of the polygon then represent the orbital energies of the [N]annulene/annulenyl system.[11] Related mnemonics exists for linear and Möbius systems.[12]
The values of α and β
The value of α is the energy of an electron in a 2p orbital, relative to an unbound electron at infinity. This quantity is negative, since the electron is stabilized by being electrostatically bound to the positively charged nucleus. For carbon this value is known to be approximately –11.4 eV. Since Hückel theory is generally only interested in energies relative to a reference localized system, the value of α is often immaterial and can be set to zero without affecting any conclusions.
Roughly speaking, β physically represents the energy of stabilization experienced by an electron allowed to delocalize in a π molecular orbital formed from the 2p orbitals of adjacent atoms, compared to being localized in an isolated 2p atomic orbital. As such, it is also a negative number, although it is often spoken of in terms of its absolute value. The value for |β| in Hückel theory is roughly constant for structurally similar compounds, but not surprisingly, structurally dissimilar compounds will give very different values for |β|. For example, using the π bond energy of ethylene (65 kcal/mole) and comparing the energy of a doubly-occupied π orbital (2α + 2β) with the energy of electrons in two isolated p orbitals (2α), a value of |β| = 32.5 kcal/mole can be inferred. On the other hand, using the resonance energy of benzene (36 kcal/mole, derived from heats of hydrogenation) and comparing benzene (6α + 8β) with a hypothetical "non-aromatic 1,3,5-cyclohexatriene" (6α + 6β), a much smaller value of |β| = 18 kcal/mole emerges. These differences are not surprising, given the substantially shorter bond length of ethylene (1.33 Å) compared to benzene (1.40 Å). The shorter distance between the interacting p orbitals accounts for the greater energy of interaction, which is reflected by a higher value of |β|. Nevertheless, heat of hydrogenation measurements of various polycyclic aromatic hydrocarbons like naphthalene and anthracene all imply values of |β| between 17 and 20 kcal/mol.
However, even for the same compound, the correct assignment of |β| can be controversial. For instance, it is argued that the resonance energy measured experimentally via heats of hydrogenation is diminished by the distortions in bond lengths that must take place going from the single and double bonds of "non-aromatic 1,3,5-cyclohexatriene" to the delocalized bonds of benzene. Taking this distortion energy into account, the value of |β| for delocalization without geometric change (called the "vertical resonance energy") for benzene is found to be around 37 kcal/mole. On the other hand, experimental measurements of electronic spectra have given a value of |β| (called the "spectroscopic resonance energy") as high as 3 eV (~70 kcal/mole) for benzene.[13] Given these subtleties, qualifications, and ambiguities, Hückel theory should not be called upon to provide accurate quantitative predictions – only semi-quantitative or qualitative trends and comparisons are reliable and robust.
Other successful predictions
With this caveat in mind, many predictions of the theory have been experimentally verified:
- The HOMO–LUMO gap, in terms of the β constant, correlates directly with the respective molecular electronic transitions observed with UV/VIS spectroscopy. For linear polyenes, the energy gap is given as:

- The predicted molecular orbital energies as stipulated by Koopmans' theorem correlate with photoelectron spectroscopy.[15]
- The Hückel delocalization energy correlates with the experimental heat of combustion. This energy is defined as the difference between the total predicted π energy (in benzene 8β) and a hypothetical π energy in which all ethylene units are assumed isolated, each contributing 2β (making benzene 3 × 2β = 6β).
- Molecules with molecular orbitals paired up such that only the sign differs (for example α ± β) are called alternant hydrocarbons and have in common small molecular dipole moments. This is in contrast to non-alternant hydrocarbons, such as azulene and fulvene that have large dipole moments. The Hückel theory is more accurate for alternant hydrocarbons.
- For cyclobutadiene the theory predicts that the two high-energy electrons occupy a degenerate pair of molecular orbitals (following from Hund's rules) that are neither stabilized nor destabilized. Hence the square molecule would be a very reactive triplet diradical (the ground state is actually rectangular without degenerate orbitals). In fact, all cyclic conjugated hydrocarbons with a total of 4n π-electrons share this molecular orbital pattern, and this forms the basis of Hückel's rule.
- Dewar reactivity numbers deriving from the Hückel approach correctly predict the reactivity of aromatic systems with nucleophiles and electrophiles.
- The benzyl cation and anion serve as simple models for arenes with electron-withdrawing and electron-donating groups, respectively. The π-electron population correctly implies the meta- and ortho-/para-selectivity for electrophilic aromatic substitution of π electron-poor and π electron-rich arenes, respectively.
Application in optical activity analysis
The analysis of the optical activity of a molecule depends to a certain extent on the study of its chiral characteristics. However, for achiral molecules applying pesudoscalars to simplify the calculations of optical activity cannot be achieved due to the lack of spatial average.[16]
Instead of traditional chiroptical solution measurements, Hückel theory helps focus on oriented π systems by separating from σ electrons especially in the planar, -symmetric cases. Transition dipole moments derived by multiplying each wavefunction of individual planar molecule one by one, contribute to the directions of the most optical activity, where sit at the bisectors of two orthogonal ones. Despite the zero value for the trace of the tensor, cis-butadiene shows considerable off diagonal component which was computed as the first optical activity evaluation of achiral molecule.[17]

By using 3,5-dimethylene-1-cyclopentene as an example. Transition electric dipole, magnetic dipole and electric quadrupole moments interactions result in optical rotation(OR), which can be described by both tensor components and chemical geometries. The in phase overlap of two molecular orbitals yield negative charge while depleting charge out of phase. The movement can be interpreted quantitatively by corresponding π and π* orbitals coefficients.
Delocalization energy, π-bond orders, and π-electron populations
The delocalization energy, π-bond orders, and π-electron population are chemically significant parameters that can be gleaned from the orbital energies and coefficients that are the direct outputs of Hückel theory.[18] These are quantities strictly derived from theory, as opposed to measurable physical properties, though they correlate with measurable qualitative and quantitative properties of the chemical species. Delocalization energy is defined as the difference in energy between that of the most stable localized Lewis structure and the energy of the molecule computed from Hückel theory orbital energies and occupancies. Since all energies are relative, we set without loss of generality to simplify discussion. The energy of the localized structure is then set to be 2β for every two-electron localized π-bond. The Hückel energy of the molecule is , where the sum is over all Hückel orbitals, is the occupancy of orbital i, set to be 2 for doubly-occupied orbitals, 1 for singly-occupied orbitals, and 0 for unoccupied orbitals, and is the energy of orbital i. Thus, the delocalization energy, conventionally a positive number, is defined as




- .
 π-Electron populations of benzyl cation and benzyl anion can be used to rationalize the directing group effects of electron-withdrawing and -donating substituents in electrophilic aromatic substitution.
π-Electron populations of benzyl cation and benzyl anion can be used to rationalize the directing group effects of electron-withdrawing and -donating substituents in electrophilic aromatic substitution.

In the case of benzene, the occupied orbitals have energies (again setting ) 2β, β, and β. This gives the Hückel energy of benzene as . Each Kekulé structure of benzene has three double bonds, so the localized structure is assigned an energy of . The delocalization energy, measured in units of , is then .




The π-bond orders derived from Hückel theory are defined using the orbital coefficients of the Hückel MOs. The π-bond order between atoms j and k is defined as
- ,

where is again the orbital occupancy of orbital i and and are the coefficients on atoms j and k, respectively, for orbital i. For benzene, the three occupied MOs, expressed as linear combinations of AOs , are:[19]



- , [];
- , [];
- , [].





Perhaps surprisingly, the π-bond order formula gives a bond order of

for the bond between carbons 1 and 2.[20] The resulting total (σ + π) bond order of is the same between any other pair of adjacent carbon atoms. This is more than the naive π-bond order of (for a total bond order of ) that one might guess when simply considering the Kekulé structures and the usual definition of bond order in valence bond theory. The Hückel definition of bond order attempts to quantify any additional stabilization that the system enjoys resulting from delocalization. In a sense, the Hückel bond order suggests that there are four π-bonds in benzene instead of the three that are implied by the Kekulé-type Lewis structures. The "extra" bond is attributed to the additional stabilization that results from the aromaticity of the benzene molecule. (This is only one of several definitions for non-integral bond orders, and other definitions will lead to different values that fall between 1 and 2.)



The π-electron population is calculated in a very similar way to the bond order using the orbital coefficients of the Hückel MOs. The π-electron population on atom j is defined as
- .
![{\displaystyle n_{\pi }(j)=\sum _{i}n_{i}[c_{j}^{(i)}]^{2}}](../I/959cf318c1d2bbcc25cf3e622c9c86af05d21344.svg)
The associated Hückel Coulomb charge is defined as , where is the number of π-electrons contributed by a neutral, sp2-hybridized atom j (we always have for carbon).



For carbon 1 on benzene, this yields a π-electron population of
- .

Since each carbon atom contributes one π-electron to the molecule, this gives a Coulomb charge of 0 for carbon 1 (and all other carbon atoms), as expected.
In the cases of benzyl cation and benzyl anion shown above,
- and ,
- and .




Mathematics behind the Hückel method
The mathematics of the Hückel method is based on the Ritz method. In short, given a basis set of n normalized atomic orbitals , an ansatz molecular orbital is written down, with normalization constant N and coefficients which are to be determined. In other words, we are assuming that the molecular orbital (MO) can be written as a linear combination of atomic orbitals, a conceptually intuitive and convenient approximation (the linear combination of atomic orbitals or LCAO approximation). The variational theorem states that given an eigenvalue problem with smallest eigenvalue and corresponding wavefunction , any normalized trial wavefunction (i.e., holds) will satisfy








- ,
![{\displaystyle {\mathcal {E}}[\psi _{g}]=\langle \psi _{g}|{\hat {H}}|\psi _{g}\rangle =\int _{\mathbb {R} ^{3}}\psi _{g}^{*}\,{\hat {H}}\psi _{g}\,dV\geq E^{(0)}}](../I/882d4a0bf384ef760c73647f6656daf4401f9926.svg)
with equality holding if and only if . Thus, by minimizing with respect to coefficients for normalized trial wavefunctions , we obtain a closer approximation of the true ground-state wavefunction and its energy.

![{\displaystyle E(c_{1},\ldots ,c_{n})={\mathcal {E}}[\psi _{g}]}](../I/38486a6c2d3a8996ef838590f6e9f8ad37fb94e1.svg)

To start, we apply the normalization condition to the ansatz and expand to get an expression for N in terms of the . Then, we substitute the ansatz into the expression for E and expand, yielding
- , where ,
- , and .
![{\displaystyle E(c_{1},\ldots ,c_{n})=N^{2}{\Big [}\sum _{i=1}^{n}c_{i}^{2}H_{ii}+\sum _{1\leq i\neq j\leq n}c_{i}c_{j}H_{ij}{\Big ]}}](../I/5daa08b532f11bb827420d38840f2915b1f865b5.svg)
![{\displaystyle N={\Big [}\sum _{i=1}^{n}c_{i}^{2}S_{ii}+\sum _{1\leq i\neq j\leq n}c_{i}c_{j}S_{ij}{\Big ]}^{-1/2}}](../I/62d47be3fa20635a02aae4086496f33aa27b879f.svg)


In the remainder of the derivation, we will assume that the atomic orbitals are real. (For the simple case of the Hückel theory, they will be the 2pz orbitals on carbon.) Thus, , and because the Hamiltonian operator is hermitian, . Setting for to minimize E and collecting terms, we obtain a system of n simultaneous equations




- .

When , and are called the overlap and resonance (or exchange) integrals, respectively, while is called the Coulomb integral, and simply expresses the fact that the are normalized. The n × n matrices and are known as the overlap and Hamiltonian matrices, respectively.





![{\displaystyle [S_{ij}]}](../I/c45fc4fb8be331b628a1477ac0af67df50c07c28.svg)
![{\displaystyle [H_{ij}]}](../I/265712cf0e865fa3b550f2d0fe2b5e894ead82d7.svg)
By a well-known result from linear algebra, nontrivial solutions to the above system of linear equations can only exist if the coefficient matrix is singular. Hence, must have a value such that the determinant of the coefficient matrix vanishes:

![{\displaystyle [H_{ij}-ES_{ij}]}](../I/e651f73470d0025ee14b87a6857d261441d352bf.svg)

- . (*)
![{\displaystyle \mathrm {det} ([H_{ij}-ES_{ij}])=0}](../I/0bfbae02589560469d899601aa7e212b72f95f78.svg)
This determinant expression is known as the secular determinant and gives rise to a generalized eigenvalue problem. The variational theorem guarantees that the lowest value of that gives rise to a nontrivial (that is, not all zero) solution vector represents the best LCAO approximation of the energy of the most stable π orbital; higher values of with nontrivial solution vectors represent reasonable estimates of the energies of the remaining π orbitals.
The Hückel method makes a few further simplifying assumptions concerning the values of the and . In particular, it is first assumed that distinct have zero overlap. Together with the assumption that are normalized, this means that the overlap matrix is the n × n identity matrix: . Solving for E in (*) then reduces to finding the eigenvalues of the Hamiltonian matrix.
![{\displaystyle [S_{ij}]=\mathbf {I} _{n}}](../I/3f18c42631ff80e8edab8c89a5df99468db788ac.svg)
Second, in the simplest case of a planar, unsaturated hydrocarbon, the Hamiltonian matrix is parameterized in the following way:
![{\displaystyle \mathbf {H} =[H_{ij}]}](../I/0946fe29c123ab304555d70a920dcd059e108bfc.svg)
- (**)

To summarize, we are assuming that: (1) the energy of an electron in an isolated C(2pz) orbital is ; (2) the energy of interaction between C(2pz) orbitals on adjacent carbons i and j (i.e., i and j are connected by a σ-bond) is ; (3) orbitals on carbons not joined in this way are assumed not to interact, so for nonadjacent i and j; and, as mentioned above, (4) the spatial overlap of electron density between different orbitals, represented by non-diagonal elements of the overlap matrix, is ignored by setting , even when the orbitals are adjacent.




This neglect of orbital overlap is an especially severe approximation. In actuality, orbital overlap is a prerequisite for orbital interaction, and it is impossible to have while . For typical bond distances (1.40 Å) as might be found in benzene, for example, the true value of the overlap for C(2pz) orbitals on adjacent atoms i and j is about ; even larger values are found when the bond distance is shorter (e.g., ethylene).[21] A major consequence of having nonzero overlap integrals is the fact that, compared to non-interacting isolated orbitals, bonding orbitals are not energetically stabilized by nearly as much as antibonding orbitals are destabilized. The orbital energies derived from the Hückel treatment do not account for this asymmetry (see Hückel solution for ethylene (below) for details).



The eigenvalues of are the Hückel molecular orbital energies , expressed in terms of and , while the eigenvectors are the Hückel MOs , expressed as linear combinations of the atomic orbitals . Using the expression for the normalization constant N and the fact that , we can find the normalized MOs by incorporating the additional condition





- .

The Hückel MOs are thus uniquely determined when eigenvalues are all distinct. When an eigenvalue is degenerate (two or more of the are equal), the eigenspace corresponding to the degenerate energy level has dimension greater than 1, and the normalized MOs at that energy level are then not uniquely determined. When that happens, further assumptions pertaining to the coefficients of the degenerate orbitals (usually ones that make the MOs orthogonal and mathematically convenient[22]) have to be made in order to generate a concrete set of molecular orbital functions.
If the substance is a planar, unsaturated hydrocarbon, the coefficients of the MOs can be found without appeal to empirical parameters, while orbital energies are given in terms of only and . On the other hand, for systems containing heteroatoms, such as pyridine or formaldehyde, values of correction constants and have to be specified for the atoms and bonds in question, and and in (**) are replaced by and , respectively.




Hückel solution for ethylene in detail
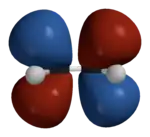

In the Hückel treatment for ethylene, we write the Hückel MOs as a linear combination of the atomic orbitals (2p orbitals) on each of the carbon atoms:

- .



Applying the result obtained by the Ritz method, we have the system of equations
- ,

where:
- and
- .


(Since 2pz atomic orbital can be expressed as a pure real function, the * representing complex conjugation can be dropped.) The Hückel method assumes that all overlap integrals (including the normalization integrals) equal the Kronecker delta, , all Coulomb integrals are equal, and the resonance integral is nonzero when the atoms i and j are bonded. Using the standard Hückel variable names, we set



- ,
- ,
- , and
- .




The Hamiltonian matrix is
- .

The matrix equation that needs to be solved is then
- ,

or, dividing by ,
- .

Setting , we obtain

- . (***)

This homogeneous system of equations has nontrivial solutions for (solutions besides the physically meaningless ) iff the matrix is singular and the determinant is zero:


- .

Solving for ,

- , or
- .


Since , the energy levels are

- , or
- .


The coefficients can then be found by expanding (***):
- and
- .


Since the matrix is singular, the two equations are linearly dependent, and the solution set is not uniquely determined until we apply the normalization condition. We can only solve for in terms of :


- , or
- .


After normalization with , the numerical values of and can be found:

- and .


Finally, the Hückel molecular orbitals are
- .

The constant β in the energy term is negative; therefore, with is the lower energy corresponding to the HOMO energy and with is the LUMO energy.




If, contrary to the Hückel treatment, a positive value for were included, the energies would instead be

- ,

while the corresponding orbitals would take the form
- .

An important consequence of setting is that the bonding (in-phase) combination is always stabilized to a lesser extent than the antibonding (out-of-phase) combination is destabilized, relative to the energy of the free 2p orbital. Thus, in general, 2-center 4-electron interactions, where both the bonding and antibonding orbitals are occupied, are destabilizing overall. This asymmetry is ignored by Hückel theory. In general, for the orbital energies derived from Hückel theory, the sum of stabilization energies for the bonding orbitals is equal to the sum of destabilization energies for the antibonding orbitals, as in the simplest case of ethylene shown here and the case of butadiene shown below.

Hückel solution for 1,3-butadiene
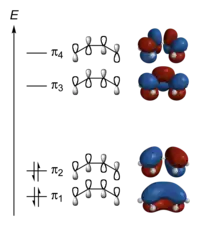
The Hückel MO theory treatment of 1,3-butadiene is largely analogous to the treatment of ethylene, shown in detail above, though we must now find the eigenvalues and eigenvectors of a 4 × 4 Hamiltonian matrix. We first write the molecular orbital as a linear combination of the four atomic orbitals (carbon 2p orbitals) with coefficients :
- .

The Hamiltonian matrix is
- .

In the same way, we write the secular equations in matrix form as
- ,

which leads to

and
- , or approximately,
- , where 1.618... and 0.618... are the golden ratios and .




The orbitals are given by
- ,
- ,
- , and
- .




See also
External links
- "Hückel method" at chem.swin.edu.au, webpage: mod3-huckel.
- N. Goudard; Y. Carissan; D. Hagebaum-Reignier; S. Humbel (2008). "HuLiS : Java Applet – Simple Hückel Theory and Mesomery – program logiciel software" (in French). Retrieved 19 August 2010.
- Rauk, Arvi. SHMO, Simple Hückel Molecular Orbital Theory Calculator. Java Applet (downloadable) Archived 2018-06-22 at the Wayback Machine.
Further reading
- The HMO-Model and its applications: Basis and Manipulation, E. Heilbronner and H. Bock, English translation, 1976, Verlag Chemie.
- The HMO-Model and its applications: Problems with Solutions, E. Heilbronner and H. Bock, English translation, 1976, Verlag Chemie.
- The HMO-Model and its applications: Tables of Hückel Molecular Orbitals, E. Heilbronner and H. Bock, English translation, 1976, Verlag Chemie.
References
- ↑ E. Hückel, Zeitschrift für Physik, 70, 204 (1931); 72, 310 (1931); 76, 628 (1932); 83, 632 (1933).
- ↑ Hückel Theory for Organic Chemists, C. A. Coulson, B. O'Leary and R. B. Mallion, Academic Press, 1978.
- ↑ P. R. Bunker and P. Jensen (2005),Fundamentals of Molecular Symmetry (CRC Press) Hückel method for benzene: Sections 3.4.3 and 10.2 ISBN 0-7503-0941-5
- ↑ Andrew Streitwieser, Molecular Orbital Theory for Organic Chemists, Wiley, New York (1961).
- ↑ "Stereochemistry of Electrocyclic Reactions", R. B. Woodward, Roald Hoffmann, J. Am. Chem. Soc., 1965; 87(2); 395–397. doi:10.1021/ja01080a054.
- ↑ Morris, Peter J. T.; Hornix, Willem J.; Bud, Robert; Morris, Peter J. T. (1992). "The Technology: Science Interaction: Walter Reppe and Cyclooctatetraene Chemistry". The British Journal for the History of Science. 25 (1): 145–167. doi:10.1017/S0007087400045374. JSTOR 4027009. S2CID 145124799.
- ↑ The actual ground state of cyclobutadiene is D2h (rectangular) with non-degenerate E2 (HOMO) and E3 (LUMO).
- ↑ The chemical bond, 2nd ed., J.N. Murrel, S.F.A. Kettle, J.M. Tedder, ISBN 0-471-90760-X
- ↑ Quantum Mechanics for Organic Chemists. Zimmerman, H., Academic Press, New York, 1975.
- ↑ Due to the twisted geometry required for a molecule to take on Möbius aromaticity, the idealized Möbius versions of the annulenes (or annulenyl ions) are hypothetical species for small ring sizes. Three- to seven-membered Möbius annulene/annulenyl systems are too twisted to be reasonably considered as stable species. Computations have considered whether certain Möbius topology isomers and conformers of eight-membered and larger annulene/annulenyl systems are aromatic. While Möbius aromatic configurations have indeed been found, they are still generally less stable than their nonaromatic counterparts. The sole exception appears to be penta-trans-[13]annulenyl cation, whose ground state is believed to be Möbius aromatic (Herges and coworkers, Org. Lett. 2010, 12, 1708). In fact, there are only a handful of ground state species that are thought to be Möbius aromatic. However, Möbius aromaticity is conceptually important, as many pericyclic transition states take on Möbius aromatic character, and the distinction between Hückel and Möbius topologies forms the basis of the Dewar-Zimmerman approach to the generalized pericyclic selection rules (Woodward-Hoffmann rules).
- ↑ Frost, A. A.; Musulin, B. (1953). "Mnemonic device for molecular-orbital energies". J. Chem. Phys. 21 (3): 572–573. Bibcode:1953JChPh..21..572F. doi:10.1063/1.1698970.
- ↑ Brown, A.D.; Brown, M. D. (1984). "A geometric method for determining the Huckel molecular orbital energy levels of open chain, fully conjugated molecules". J. Chem. Educ. 61 (9): 770. Bibcode:1984JChEd..61..770B. doi:10.1021/ed061p770.
- ↑ Cotton, F. Albert (1990). Chemical Applications of Group Theory (3rd ed.). New York: Wiley. pp. 438-440. ISBN 978-0471510949.
- ↑ "Use of Huckel Molecular Orbital Theory in Interpreting the Visible Spectra of Polymethine Dyes: An Undergraduate Physical Chemistry Experiment". Bahnick, Donald A., J. Chem. Educ. 1994, 71, 171.
- ↑ Huckel theory and photoelectron spectroscopy. von Nagy-Felsobuki, Ellak I. J. Chem. Educ. 1989, 66, 821.
- ↑ Murphy, Veronica L.; Kahr, Bart (22 April 2015). "Hückel Theory and Optical Activity". Journal of the American Chemical Society. 137 (15): 5177–5183. doi:10.1021/jacs.5b01763. PMID 25798796.
- ↑ Hansen, Aage E.; Bak, Keld L. (December 2000). "Ab Initio Calculations and Display of Enantiomeric and Nonenantiomeric Anisotropic Circular Dichroism: The Lowest π → π* Excitation in Butadiene, Cyclohexadiene, and Methyl-Substituted Cyclohexadienes †". The Journal of Physical Chemistry A. 104 (48): 11362–11370. Bibcode:2000JPCA..10411362H. doi:10.1021/jp001899+.
- ↑ Levine, Ira N. (2000). Quantum Chemistry (5th ed.). Upper Saddle River, N. J.: Prentice Hall. pp. 629-649. ISBN 0-13-685512-1.
- ↑ The "canonical" representatives of the doubly degenerate E1g orbitals (with nodal planes through the x and y axes) are shown here.
- ↑ Rauk, Arvi (2001). Orbital Interactions in Organic Chemistry (2nd ed.). New York: Wiley. pp. 92. ISBN 0-471-35833-9.
- ↑ Carroll, Felix A. (2010). Perspectives on Structure and Mechanism in Organic Chemistry (2nd ed.). Hoboken, N.J.: Wiley. p. 179. ISBN 978-0-470-27610-5.
- ↑ Strictly speaking, the only requirement is for the coefficients to be chosen so that linear combinations of the degenerate MOs span the eigenspace corresponding to that eigenvalue (energy level).