Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.
Application
Physical organic chemists use theoretical and experimental approaches work to understand these foundational problems in organic chemistry, including classical and statistical thermodynamic calculations, quantum mechanical theory and computational chemistry, as well as experimental spectroscopy (e.g., NMR), spectrometry (e.g., MS), and crystallography approaches. The field therefore has applications to a wide variety of more specialized fields, including electro- and photochemistry, polymer and supramolecular chemistry, and bioorganic chemistry, enzymology, and chemical biology, as well as to commercial enterprises involving process chemistry, chemical engineering, materials science and nanotechnology, and pharmacology in drug discovery by design.
Scope
Physical organic chemistry is the study of the relationship between structure and reactivity of organic molecules. More specifically, physical organic chemistry applies the experimental tools of physical chemistry to the study of the structure of organic molecules and provides a theoretical framework that interprets how structure influences both mechanisms and rates of organic reactions. It can be thought of as a subfield that bridges organic chemistry with physical chemistry.
Physical organic chemists use both experimental and theoretical disciplines such as spectroscopy, spectrometry, crystallography, computational chemistry, and quantum theory to study both the rates of organic reactions and the relative chemical stability of the starting materials, transition states, and products.[1] Chemists in this field work to understand the physical underpinnings of modern organic chemistry, and therefore physical organic chemistry has applications in specialized areas including polymer chemistry, supramolecular chemistry, electrochemistry, and photochemistry.[1]
History
The term physical organic chemistry was itself coined by Louis Hammett in 1940 when he used the phrase as a title for his textbook.[2]
Chemical structure and thermodynamics
Thermochemistry
Organic chemists use the tools of thermodynamics to study the bonding, stability, and energetics of chemical systems. This includes experiments to measure or determine the enthalpy (ΔH), entropy (ΔS), and Gibbs' free energy (ΔG) of a reaction, transformation, or isomerization. Chemists may use various chemical and mathematical analyses, such as a Van 't Hoff plot, to calculate these values.
Empirical constants such as bond dissociation energy, standard heat of formation (ΔHf°), and heat of combustion (ΔHc°) are used to predict the stability of molecules and the change in enthalpy (ΔH) through the course of the reactions. For complex molecules, a ΔHf° value may not be available but can be estimated using molecular fragments with known heats of formation. This type of analysis is often referred to as Benson group increment theory, after chemist Sidney Benson who spent a career developing the concept.[1] [3][4]
The thermochemistry of reactive intermediates—carbocations, carbanions, and radicals—is also of interest to physical organic chemists. Group increment data are available for radical systems.[1] Carbocation and carbanion stabilities can be assessed using hydride ion affinities and pKa values, respectively.[1]
Conformational analysis
One of the primary methods for evaluating chemical stability and energetics is conformational analysis. Physical organic chemists use conformational analysis to evaluate the various types of strain present in a molecule to predict reaction products.[5] Strain can be found in both acyclic and cyclic molecules, manifesting itself in diverse systems as torsional strain, allylic strain, ring strain, and syn-pentane strain.[1] A-values provide a quantitative basis for predicting the conformation of a substituted cyclohexane, an important class of cyclic organic compounds whose reactivity is strongly guided by conformational effects. The A-value is the difference in the Gibbs' free energy between the axial and equatorial forms of substituted cyclohexane, and by adding together the A-values of various substituents it is possible to quantitatively predict the preferred conformation of a cyclohexane derivative.
In addition to molecular stability, conformational analysis is used to predict reaction products. One commonly cited example of the use of conformational analysis is a bi-molecular elimination reaction (E2). This reaction proceeds most readily when the nucleophile attacks the species that is antiperiplanar to the leaving group. A molecular orbital analysis of this phenomenon suggest that this conformation provides the best overlap between the electrons in the R-H σ bonding orbital that is undergoing nucleophilic attack and the empty σ* antibonding orbital of the R-X bond that is being broken.[6] By exploiting this effect, conformational analysis can be used to design molecules that possess enhanced reactivity.
The physical processes which give rise to bond rotation barriers are complex, and these barriers have been extensively studied through experimental and theoretical methods.[7][8][9] A number of recent articles have investigated the predominance of the steric, electrostatic, and hyperconjugative contributions to rotational barriers in ethane, butane, and more substituted molecules.[10]
Non-covalent interactions
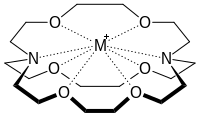
Chemists use the study of intramolecular and intermolecular non-covalent bonding/interactions in molecules to evaluate reactivity. Such interactions include, but are not limited to, hydrogen bonding, electrostatic interactions between charged molecules, dipole-dipole interactions, polar-π and cation-π interactions, π-stacking, donor-acceptor chemistry, and halogen bonding. In addition, the hydrophobic effect—the association of organic compounds in water—is an electrostatic, non-covalent interaction of interest to chemists. The precise physical origin of the hydrophobic effect originates from many complex interactions, but it is believed to be the most important component of biomolecular recognition in water.[1] For example, Xu and Melcher et al. elucidated the structural basis for folic acid recognition by folate acid receptor proteins.[11] The strong interaction between folic acid and folate receptor was attributed to both hydrogen bonds and hydrophobic interactions. The study of non-covalent interactions is also used to study binding and cooperativity in supramolecular assemblies and macrocyclic compounds such as crown ethers and cryptands, which can act as hosts to guest molecules.
Acid–base chemistry

The properties of acids and bases are relevant to physical organic chemistry. Organic chemists are primarily concerned with Brønsted–Lowry acids/bases as proton donors/acceptors and Lewis acids/bases as electron acceptors/donors in organic reactions. Chemists use a series of factors developed from physical chemistry -- electronegativity/Induction, bond strengths, resonance, hybridization, aromaticity, and solvation—to predict relative acidities and basicities.
The hard/soft acid/base principle is utilized to predict molecular interactions and reaction direction. In general, interactions between molecules of the same type are preferred. That is, hard acids will associate with hard bases, and soft acids with soft bases. The concept of hard acids and bases is often exploited in the synthesis of inorganic coordination complexes.
Kinetics
Physical organic chemists use the mathematical foundation of chemical kinetics to study the rates of reactions and reaction mechanisms. Unlike thermodynamics, which is concerned with the relative stabilities of the products and reactants (ΔG°) and their equilibrium concentrations, the study of kinetics focuses on the free energy of activation (ΔG‡) -- the difference in free energy between the reactant structure and the transition state structure—of a reaction, and therefore allows a chemist to study the process of equilibration.[1] Mathematically derived formalisms such as the Hammond Postulate, the Curtin-Hammett principle, and the theory of microscopic reversibility are often applied to organic chemistry. Chemists have also used the principle of thermodynamic versus kinetic control to influence reaction products.
Rate laws
The study of chemical kinetics is used to determine the rate law for a reaction. The rate law provides a quantitative relationship between the rate of a chemical reaction and the concentrations or pressures of the chemical species present.[12] Rate laws must be determined by experimental measurement and generally cannot be elucidated from the chemical equation. The experimentally determined rate law refers to the stoichiometry of the transition state structure relative to the ground state structure. Determination of the rate law was historically accomplished by monitoring the concentration of a reactant during a reaction through gravimetric analysis, but today it is almost exclusively done through fast and unambiguous spectroscopic techniques. In most cases, the determination of rate equations is simplified by adding a large excess ("flooding") all but one of the reactants.
Catalysis

The study of catalysis and catalytic reactions is very important to the field of physical organic chemistry. A catalyst participates in the chemical reaction but is not consumed in the process.[12] A catalyst lowers the activation energy barrier (ΔG‡), increasing the rate of a reaction by either stabilizing the transition state structure or destabilizing a key reaction intermediate, and as only a small amount of catalyst is required it can provide economic access to otherwise expensive or difficult to synthesize organic molecules. Catalysts may also influence a reaction rate by changing the mechanism of the reaction.[1]
Kinetic isotope effect
Although a rate law provides the stoichiometry of the transition state structure, it does not provide any information about breaking or forming bonds.[1] The substitution of an isotope near a reactive position often leads to a change in the rate of a reaction. Isotopic substitution changes the potential energy of reaction intermediates and transition states because heavier isotopes form stronger bonds with other atoms. Atomic mass affects the zero-point vibrational state of the associated molecules, shorter and stronger bonds in molecules with heavier isotopes and longer, weaker bonds in molecules with light isotopes.[6] Because vibrational motions will often change during a course of a reaction, due to the making and breaking of bonds, the frequencies will be affected, and the substitution of an isotope can provide insight into the reaction mechanism and rate law.
Substituent effects
The study of how substituents affect the reactivity of a molecule or the rate of reactions is of significant interest to chemists. Substituents can exert an effect through both steric and electronic interactions, the latter of which include resonance and inductive effects. The polarizability of molecule can also be affected. Most substituent effects are analyzed through linear free energy relationships (LFERs). The most common of these is the Hammett Plot Analysis.[1] This analysis compares the effect of various substituents on the ionization of benzoic acid with their impact on diverse chemical systems. The parameters of the Hammett plots are sigma (σ) and rho (ρ). The value of σ indicates the acidity of substituted benzoic acid relative to the unsubstituted form. A positive σ value indicates the compound is more acidic, while a negative value indicates that the substituted version is less acidic. The ρ value is a measure of the sensitivity of the reaction to the change in substituent, but only measures inductive effects. Therefore, two new scales were produced that evaluate the stabilization of localized charge through resonance. One is σ+, which concerns substituents that stabilize positive charges via resonance, and the other is σ− which is for groups that stabilize negative charges via resonance. Hammett analysis can be used to help elucidate the possible mechanisms of a reaction. For example, if it is predicted that the transition state structure has a build-up of negative charge relative to the ground state structure, then electron-donating groups would be expected to increase the rate of the reaction.[1]
Other LFER scales have been developed. Steric and polar effects are analyzed through Taft Parameters. Changing the solvent instead of the reactant can provide insight into changes in charge during the reaction. The Grunwald-Winstein Plot provides quantitative insight into these effects.[1] [13]
Solvent effects
Solvents can have a powerful effect on solubility, stability, and reaction rate. A change in solvent can also allow a chemist to influence the thermodynamic or kinetic control of the reaction. Reactions proceed at different rates in different solvents due to the change in charge distribution during a chemical transformation. Solvent effects may operate on the ground state and/or transition state structures.[1]
An example of the effect of solvent on organic reactions is seen in the comparison of SN1 and SN2 reactions.[14]
Solvent can also have a significant effect on the thermodynamic equilibrium of a system, for instance as in the case of keto-enol tautomerizations. In non-polar aprotic solvents, the enol form is strongly favored due to the formation of an intramolecular hydrogen-bond, while in polar aprotic solvents, such as methylene chloride, the enol form is less favored due to the interaction between the polar solvent and the polar diketone. In protic solvents, the equilibrium lies towards the keto form as the intramolecular hydrogen bond competes with hydrogen bonds originating from the solvent.[15] [16] [17]

A modern example of the study of solvent effects on chemical equilibrium can be seen in a study of the epimerization of chiral cyclopropylnitrile Grignard reagents.[18] This study reports that the equilibrium constant for the cis to trans isomerization of the Grignard reagent is much greater—the preference for the cis form is enhanced—in THF as a reaction solvent, over diethyl ether. However, the faster rate of cis-trans isomerization in THF results in a loss of stereochemical purity. This is a case where understanding the effect of solvent on the stability of the molecular configuration of a reagent is important with regard to the selectivity observed in an asymmetric synthesis.
Quantum chemistry
Many aspects of the structure-reactivity relationship in organic chemistry can be rationalized through resonance, electron pushing, induction, the eight electron rule, and s-p hybridization, but these are only helpful formalisms and do not represent physical reality. Due to these limitations, a true understanding of physical organic chemistry requires a more rigorous approach grounded in particle physics. Quantum chemistry provides a rigorous theoretical framework capable of predicting the properties of molecules through calculation of a molecule's electronic structure, and it has become a readily available tool in physical organic chemists in the form of popular software packages. The power of quantum chemistry is built on the wave model of the atom, in which the nucleus is a very small, positively charged sphere surrounded by a diffuse electron cloud. Particles are defined by their associated wavefunction, an equation which contains all information associated with that particle.[12] All information about the system is contained in the wavefunction. This information is extracted from the wavefunction through the use of mathematical operators.

The energy associated with a particular wavefunction, perhaps the most important information contained in a wavefunction, can be extracted by solving the Schrödinger equation (above, Ψ is the wavefunction, E is the energy, and Ĥ is the Hamiltonian operator)[12] in which an appropriate Hamiltonian operator is applied. In the various forms of the Schrödinger equation, the overall size of a particle's probability distribution increases with decreasing particle mass. For this reason, nuclei are of negligible size in relation to much lighter electrons and are treated as point charges in practical applications of quantum chemistry.
Due to complex interactions which arise from electron-electron repulsion, algebraic solutions of the Schrödinger equation are only possible for systems with one electron such as the hydrogen atom, H2+, H32+, etc.; however, from these simple models arise all the familiar atomic (s,p,d,f) and bonding (σ,π) orbitals. In systems with multiple electrons, an overall multielectron wavefunction describes all of their properties at once. Such wavefunctions are generated through the linear addition of single electron wavefunctions to generate an initial guess, which is repeatedly modified until its associated energy is minimized. Thousands of guesses are often required until a satisfactory solution is found, so such calculations are performed by powerful computers. Importantly, the solutions for atoms with multiple electrons give properties such as diameter and electronegativity which closely mirror experimental data and the patterns found in the periodic table. The solutions for molecules, such as methane, provide exact representations of their electronic structure which are unobtainable by experimental methods. Instead of four discrete σ-bonds from carbon to each hydrogen atom, theory predicts a set of four bonding molecular orbitals which are delocalized across the entire molecule. Similarly, the true electronic structure of 1,3-butadiene shows delocalized π-bonding molecular orbitals stretching through the entire molecule rather than two isolated double bonds as predicted by a simple Lewis structure.
A complete electronic structure offers great predictive power for organic transformations and dynamics, especially in cases concerning aromatic molecules, extended π systems, bonds between metal ions and organic molecules, molecules containing nonstandard heteroatoms like selenium and boron, and the conformational dynamics of large molecules such as proteins wherein the many approximations in chemical formalisms make structure and reactivity prediction impossible. An example of how electronic structure determination is a useful tool for the physical organic chemist is the metal-catalyzed dearomatization of benzene. Chromium tricarbonyl is highly electrophilic due to the withdrawal of electron density from filled chromium d-orbitals into antibonding CO orbitals, and is able to covalently bond to the face of a benzene molecule through delocalized molecular orbitals. The CO ligands inductively draw electron density from benzene through the chromium atom, and dramatically activate benzene to nucleophilic attack. Nucleophiles are then able to react to make hexacyclodienes, which can be used in further transformations such as Diels Alder cycloadditions.[19]

Quantum chemistry can also provide insight into the mechanism of an organic transformation without the collection of any experimental data. Because wavefunctions provide the total energy of a given molecular state, guessed molecular geometries can be optimized to give relaxed molecular structures very similar to those found through experimental methods.[20] Reaction coordinates can then be simulated, and transition state structures solved. Solving a complete energy surface for a given reaction is therefore possible, and such calculations have been applied to many problems in organic chemistry where kinetic data is unavailable or difficult to acquire.[1]
Spectroscopy, spectrometry, and crystallography
Physical organic chemistry often entails the identification of molecular structure, dynamics, and the concentration of reactants in the course of a reaction. The interaction of molecules with light can afford a wealth of data about such properties through nondestructive spectroscopic experiments, with light absorbed when the energy of a photon matches the difference in energy between two states in a molecule and emitted when an excited state in a molecule collapses to a lower energy state. Spectroscopic techniques are broadly classified by the type of excitation being probed, such as vibrational, rotational, electronic, nuclear magnetic resonance (NMR), and electron paramagnetic resonance spectroscopy. In addition to spectroscopic data, structure determination is often aided by complementary data collected from X-Ray diffraction and mass spectrometric experiments.[21]
NMR and EPR spectroscopy

One of the most powerful tools in physical organic chemistry is NMR spectroscopy. An external magnetic field applied to a paramagnetic nucleus generates two discrete states, with positive and negative spin values diverging in energy; the difference in energy can then be probed by determining the frequency of light needed to excite a change in spin state for a given magnetic field. Nuclei that are not indistinguishable in a given molecule absorb at different frequencies, and the integrated peak area in an NMR spectrum is proportional to the number of nuclei responding to that frequency.[22] It is possible to quantify the relative concentration of different organic molecules simply by integration peaks in the spectrum, and many kinetic experiments can be easily and quickly performed by following the progress of a reaction within one NMR sample. Proton NMR is often used by the synthetic organic chemist because protons associated with certain functional groups give characteristic absorption energies, but NMR spectroscopy can also be performed on isotopes of nitrogen, carbon, fluorine, phosphorus, boron, and a host of other elements. In addition to simple absorption experiments, it is also possible to determine the rate of fast atom exchange reactions through suppression exchange measurements, interatomic distances through multidimensional nuclear Overhauser effect experiments, and through-bond spin-spin coupling through homonuclear correlation spectroscopy.[23] In addition to the spin excitation properties of nuclei, it is also possible to study the properties of organic radicals through the same fundamental technique. Unpaired electrons also have a net spin, and an external magnetic field allows for the extraction of similar information through electron paramagnetic resonance (EPR) spectroscopy.[1]
Vibrational spectroscopy

Vibrational spectroscopy, or infrared (IR) spectroscopy, allows for the identification of functional groups and, due to its low expense and robustness, is often used in teaching labs and the real-time monitoring of reaction progress in difficult to reach environments (high pressure, high temperature, gas phase, phase boundaries). Molecular vibrations are quantized in an analogous manner to electronic wavefunctions, with integer increases in frequency leading to higher energy states. The difference in energy between vibrational states is nearly constant, often falling in the energy range corresponding to infrared photons, because at normal temperatures molecular vibrations closely resemble harmonic oscillators. It allows for the crude identification of functional groups in organic molecules, but spectra are complicated by vibrational coupling between nearby functional groups in complex molecules. Therefore, its utility in structure determination is usually limited to simple molecules. Further complicating matters is that some vibrations do not induce a change in the molecular dipole moment and will not be observable with standard IR absorption spectroscopy. These can instead be probed through Raman spectroscopy, but this technique requires a more elaborate apparatus and is less commonly performed. However, as Raman spectroscopy relies on light scattering it can be performed on microscopic samples such as the surface of a heterogeneous catalyst, a phase boundary, or on a one microliter (µL) subsample within a larger liquid volume.[21] The applications of vibrational spectroscopy are often used by astronomers to study the composition of molecular gas clouds, extrasolar planetary atmospheres, and planetary surfaces.
Electronic excitation spectroscopy
Electronic excitation spectroscopy, or ultraviolet-visible (UV-vis) spectroscopy, is performed in the visible and ultraviolet regions of the electromagnetic spectrum and is useful for probing the difference in energy between the highest energy occupied (HOMO) and lowest energy unoccupied (LUMO) molecular orbitals. This information is useful to physical organic chemists in the design of organic photochemical systems and dyes, as absorption of different wavelengths of visible light give organic molecules color. A detailed understanding of an electronic structure is therefore helpful in explaining electronic excitations, and through careful control of molecular structure it is possible to tune the HOMO-LUMO gap to give desired colors and excited state properties.[24]
Mass spectrometry
Mass spectrometry is a technique which allows for the measurement of molecular mass and offers complementary data to spectroscopic techniques for structural identification. In a typical experiment a gas phase sample of an organic material is ionized and the resulting ionic species are accelerated by an applied electric field into a magnetic field. The deflection imparted by the magnetic field, often combined with the time it takes for the molecule to reach a detector, is then used to calculate the mass of the molecule. Often in the course of sample ionization large molecules break apart, and the resulting data show a parent mass and a number of smaller fragment masses; such fragmentation can give rich insight into the sequence of proteins and nucleic acid polymers. In addition to the mass of a molecule and its fragments, the distribution of isotopic variant masses can also be determined and the qualitative presence of certain elements identified due to their characteristic natural isotope distribution. The ratio of fragment mass population to the parent ion population can be compared against a library of empirical fragmentation data and matched to a known molecular structure.[25] Combined gas chromatography and mass spectrometry is used to qualitatively identify molecules and quantitatively measure concentration with great precision and accuracy, and is widely used to test for small quantities of biomolecules and illicit narcotics in blood samples. For synthetic organic chemists it is a useful tool for the characterization of new compounds and reaction products.
Crystallography
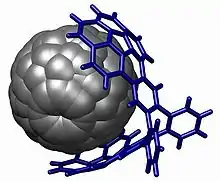
Unlike spectroscopic methods, X-ray crystallography always allows for unambiguous structure determination and provides precise bond angles and lengths totally unavailable through spectroscopy. It is often used in physical organic chemistry to provide an absolute molecular configuration and is an important tool in improving the synthesis of a pure enantiomeric substance. It is also the only way to identify the position and bonding of elements that lack an NMR active nucleus such as oxygen. Indeed, before x-ray structural determination methods were made available in the early 20th century all organic structures were entirely conjectural: tetrahedral carbon, for example, was only confirmed by the crystal structure of diamond,[26] and the delocalized structure of benzene was confirmed by the crystal structure of hexamethylbenzene.[27] While crystallography provides organic chemists with highly satisfying data, it is not an everyday technique in organic chemistry because a perfect single crystal of a target compound must be grown. Only complex molecules, for which NMR data cannot be unambiguously interpreted, require this technique. In the example below, the structure of the host–guest complex would have been quite difficult to solve without a single crystal structure: there are no protons on the fullerene, and with no covalent bonds between the two halves of the organic complex spectroscopy alone was unable to prove the hypothesized structure.
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Dougherty, Dennis A.; Anslyn, Eric V. (2006). Modern Physical Organic Chemistry. Sausalito, CA, USA: University Science Books. ISBN 9781891389313.
- ↑ Taft, R W; Deno, N C; Skell, P S (October 1958). "Physical Organic Chemistry". Annual Review of Physical Chemistry. 9 (1): 287–314. Bibcode:1958ARPC....9..287T. doi:10.1146/annurev.pc.09.100158.001443. ISSN 0066-426X.
- ↑ Cohen, N.; Benson, S. W. (1 November 1993). "Estimation of heats of formation of organic compounds by additivity methods". Chemical Reviews. 93 (7): 2419–2438. doi:10.1021/cr00023a005.
- ↑ Benson, Sidney W.; Cruickshank, F. R.; Golden, D. M.; Haugen, Gilbert R.; O'Neal, H. E.; Rodgers, A. S.; Shaw, Robert; Walsh, R. (1 June 1969). "Additivity rules for the estimation of thermochemical properties". Chemical Reviews. 69 (3): 279–324. doi:10.1021/cr60259a002.
- ↑ Carey, Francis A. (2008). Organic Chemistry (7th ed.). Boston, MA, USA: McGraw-Hill. ISBN 9780073047874.
- 1 2 Isaacs, Neil S. (1995). Physical Organic Chemistry (2nd ed.). Harlow, ESS, ENG: Longman Scientific & Technical. ISBN 978-0582218635.
- ↑ Mo, Yirong; Gao, Jiali (1 February 2007). "Theoretical Analysis of the Rotational Barrier of Ethane". Accounts of Chemical Research. 40 (2): 113–119. doi:10.1021/ar068073w. PMID 17309192. S2CID 16332261.
- ↑ Liu, Shubin (7 February 2013). "Origin and Nature of Bond Rotation Barriers: A Unified View". The Journal of Physical Chemistry A. 117 (5): 962–965. Bibcode:2013JPCA..117..962L. doi:10.1021/jp312521z. PMID 23327680.
- ↑ Liu, Shubin; Govind, Niranjan (1 July 2008). "Toward Understanding the Nature of Internal Rotation Barriers with a New Energy Partition Scheme: Ethane and-Butane". The Journal of Physical Chemistry A. 112 (29): 6690–6699. Bibcode:2008JPCA..112.6690L. doi:10.1021/jp800376a. PMID 18563887.
- ↑ Yamamoto, Takuhei; Chen, Pi-Yu; Lin, Guangxin; Błoch-Mechkour, Anna; Jacobsen, Neil E.; Bally, Thomas; Glass, Richard S. (1 October 2012). "Synthesis and rotation barriers in 2, 6-Di-(-anisyl) anisole" (PDF). Journal of Physical Organic Chemistry. 25 (10): 878–882. doi:10.1002/poc.2939.
- ↑ Chen, Chen; Ke, Jiyuan; Zhou, X. Edward; Yi, Wei; Brunzelle, Joseph S.; Li, Jun; Yong, Eu-Leong; Xu, H. Eric; Melcher, Karsten (14 July 2013). "Structural basis for molecular recognition of folic acid by folate receptors". Nature. 500 (7463): 486–489. Bibcode:2013Natur.500..486C. doi:10.1038/nature12327. PMC 5797940. PMID 23851396.
- 1 2 3 4 McQuarrie, Donald A.; Simon, John D. (1997). Physical Chemistry: A Molecular Approach (Rev. ed.). Sausalito, CA, USA: University Science Books. ISBN 9780935702996. Retrieved 21 June 2015. Note, Amazon rather than Google allows access into this text.
- ↑ Kevill, Dennis N.; D'Souza, Malcolm J. (1 June 1992). "Concerning the development of scales of solvent ionizing power based on solvolyses of benzylic substrates". Journal of Physical Organic Chemistry. 5 (6): 287–294. doi:10.1002/poc.610050602.
- ↑ Reichardt, Christian; Welton, Thomas (2011). Solvents and solvent effects in organic chemistry (4th, updated and enl. ed.). Weinheim, Germany: Wiley-VCH. ISBN 978-3-527-32473-6.
- ↑ Mills, Sander G.; Beak, Peter (1 April 1985). "Solvent effects on keto-enol equilibria: tests of quantitative models". The Journal of Organic Chemistry. 50 (8): 1216–1224. doi:10.1021/jo00208a014.
- ↑ Emsley, John; Freeman, Neville J. (1 October 1987). "β-diketone interactions". Journal of Molecular Structure. 161 (1–2): 193–204. Bibcode:1987JMoSt.161..193E. doi:10.1016/0022-2860(87)85074-3.
- ↑ Schlund, Sebastian; Basílio Janke, Eline M.; Weisz, Klaus; Engels, Bernd (1 January 2009). "Predicting the tautomeric equilibrium of acetylacetone in solution. I. The right answer for the wrong reason?". Journal of Computational Chemistry. 31 (4): 665–70. doi:10.1002/jcc.21354. PMID 19557765. S2CID 6003410.
- 1 2 Gao, Ming; Patwardhan, Neeraj N.; Carlier, Paul R. (2013). "Stereochemical Inversion of a Cyano-Stabilized Grignard Reagent: Remarkable Effects of the Ethereal Solvent Structure and Concentration". J. Am. Chem. Soc. 135 (38): 14390–14400. doi:10.1021/ja407348s. PMID 23978216.
- ↑ Semmelhack, M. F.; Hall, H. T.; Yoshifuji, M. (September 1976). ".eta.5-Cyclohexadienyltricarbonylchromium(0) intermediates in the reaction of carbanions with .eta.6-arenetricarbonylchromium(0)". Journal of the American Chemical Society. 98 (20): 6387–6389. doi:10.1021/ja00436a056.
- ↑ Schaefer III, Henry F. (2004). Quantum Chemistry: The Development of ab initio Methods in Molecular Electronic Structure Theory. Chicago, IL, USA: R.R. Donnelly (Courier, Dover). ISBN 978-0486432465. Retrieved 21 June 2015.
- 1 2 Drago, Russell S. (1992). Physical Methods for Chemists (2nd ed.). Ft. Worth, TX, USA: Saunders. ISBN 9780030970375. Retrieved 22 June 2014.
- ↑ James Keeler. "NMR and energy levels (Ch.2)" (PDF). Understanding NMR Spectroscopy. University of California, Irvine. Retrieved 2013-10-26.
- ↑ Keeler, James (2010). Understanding NMR spectroscopy (2nd ed.). Chichester: Wiley. ISBN 978-0-470-74608-0.
- ↑ Reusch, William. "Visible and Ultraviolet Spectroscopy". Michigan State University Website. Michigan State University. Retrieved 26 October 2013.
- ↑ Adlard, edited by Alan J. Handley, Edward R. (2000). Gas chromatographic techniques and applications. Boca Raton, FL: CRC Press. p. 168. ISBN 978-0-8493-0514-6.
{{cite book}}:|first=has generic name (help)CS1 maint: multiple names: authors list (link) - ↑ Bragg, W. H.; Bragg, W. L. (July 1913). "The Structure of the Diamond". Nature. 91 (2283): 557. Bibcode:1913Natur..91..557B. doi:10.1038/091557a0. S2CID 3987932.
- ↑ Lonsdale, K. (November 1928). "The Structure of the Benzene Ring". Nature. 122 (3082): 810. Bibcode:1928Natur.122..810L. doi:10.1038/122810c0. S2CID 4105837.
Further reading
General
- Peter Atkins & Julio de Paula, 2006, "Physical chemistry," 8th Edn., New York, NY, USA:Macmillan, ISBN 0716787598, accessed 21 June 2015. [E.g., see p. 422 for a group theoretical/symmetry description of atomic orbitals contributing to bonding in methane, CH4, and pp. 390f for estimation of π-electron binding energy for 1,3-butadiene by the Hückel method.]
- Thomas H. Lowry & Kathleen Schueller Richardson, 1987, Mechanism and Theory in Organic Chemistry, 3rd Edn., New York, NY, USA:Harper & Row, ISBN 0060440848, accessed 20 June 2015. [The authoritative textbook on the subject, containing a number of appendices that provide technical details on molecular orbital theory, kinetic isotope effects, transition state theory, and radical chemistry.]
- Eric V. Anslyn & Dennis A. Dougherty, 2006, Modern Physical Organic Chemistry, Sausalito, Calif.: University Science Books, ISBN 1891389319. [A modernized and streamlined treatment with an emphasis on applications and cross-disciplinary connections.]
- Michael B. Smith & Jerry March, 2007, "March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure," 6th Ed., New York, NY, USA:Wiley & Sons, ISBN 0470084944, accessed 19 June 2015.
- Francis A. Carey & Richard J. Sundberg, 2006, "Advanced Organic Chemistry: Part A: Structure and Mechanisms," 4th Edn., New York, NY, USA:Springer Science & Business Media, ISBN 0306468565, accessed 19 June 2015.
- Hammett, Louis P. (1940) Physical Organic Chemistry, New York, NY, USA: McGraw Hill, accessed 20 June 2015.
History
- Hammond, George S. (1997). "Physical organic chemistry after 50 years: It has changed, but is it still there?" (PDF). Pure Appl. Chem. 69 (9): 1919–22. doi:10.1351/pac199769091919. S2CID 53723796. Retrieved 20 June 2015. [An outstanding starting point on the history of the field, from a critically important contributor, referencing and discussing the early Hammett text, etc.]
Thermochemistry
- L. K. Doraiswamy, 2005, "Estimation of properties of organic compounds (Ch. 3)," pp. 36–51, 118-124 (refs.), in Organic Synthesis Engineering, Oxford, Oxon, ENG:Oxford University Press, ISBN 0198025696, accessed 22 June 2015. (This book chapter surveys a very wide range of physical properties and their estimation, including the narrow list of thermochemical properties appearing in the June 2015 WP article, placing the Benson et al. method alongside many other methods. L. K. Doraiswamy is Anson Marston Distinguished Professor of Engineering at Iowa State University.)
- Irikura, Karl K.; Frurip, David J. (1998). "Computational Thermochemistry". In Irikura, Karl K.; Frurip, David J. (eds.). Computational Thermochemistry: Prediction and Estimation of Molecular Thermodynamics. ACS Symposium Series. Vol. 677. American Chemical Society. pp. 2–18. doi:10.1021/bk-1998-0677.ch001. ISBN 978-0-8412-3533-5.
Branches of chemistry | |
|---|---|
| Analytical | |
| Theoretical | |
| Physical | |
| Inorganic | |
| Organic | |
| Biological | |
| Interdisciplinarity | |
| See also | |