| Hurler-Scheie syndrome | |
|---|---|
| Other names | Mucopolysaccharidosis type I H-S |
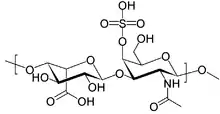 | |
| Structure of dermatan sulfate, one of the molecules that accumulates in the lysosomes of MPS I patients | |
| Usual onset | Age 3-8 years |
| Causes | Deficiency of the alpha-L iduronidase enzyme |
| Differential diagnosis | Other forms of MPS I; Hunter syndrome; other mucopolysaccharidoses |
| Treatment | Enzyme replacement therapy with iduronidase; surgery |
| Prognosis | Life expectancy is generally into the late teens or early 20s, but may vary depending on the severity of the disease |
| Frequency | 1:115,000 (Hurler-Scheie syndrome/intermediate)[1] |
Hurler–Scheie syndrome is a genetic disorder caused by the buildup of glycosaminoglycans (GAGs) in various organ tissues. It is a cutaneous condition, also characterized by mild mental retardation and corneal clouding.[2] Respiratory problems, sleep apnea, and heart disease may develop in adolescence.[1]
Hurler–Scheie syndrome is classified as a lysosomal storage disease. Patients with Hurler–Scheie syndrome lack the ability to break down GAGs in their lysosomes due a deficiency of the enzyme iduronidase.
All forms of mucopolysaccharidosis type I (MPS I) are a spectrum of the same disease. Hurler-Sheie is the subtype of MPS I with intermediate severity. Hurler syndrome is the most severe form, while Scheie syndrome is the least severe form. Some clinicians consider the differences between Hurler, Hurler-Scheie, and Scheie syndromes to be arbitrary. Instead, they classify these patients as having "severe", "intermediate", or "attenuated" MPS I.[1]
See also
References
- 1 2 3 "Mucopolysaccharidoses Fact Sheet". National Institute of Neurological Disorders and Stroke. 15 Nov 2017. Retrieved 11 May 2018.
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.