| Inborn errors of carbohydrate metabolism | |
|---|---|
| Specialty | Medical genetics |

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
An example is lactose intolerance.
Carbohydrates account for a major portion of the human diet. These carbohydrates are composed of three principal monosaccharides: glucose, fructose and galactose; in addition glycogen is the storage form of carbohydrates in humans. The failure to effectively use these molecules accounts for the majority of the inborn errors of human carbohydrates metabolism.
By Carbohydrate
Glycogen and Glucose
Glycogen storage diseases are deficiencies of enzymes or transport proteins which impair glycogen synthesis, glycogen degradation or glycolysis. The two organs most commonly affected are the liver and the skeletal muscle. Glycogen storage diseases that affect the liver typically cause hepatomegaly and hypoglycemia; those that affect skeletal muscle cause exercise intolerance, progressive weakness and cramping.[1]
Glucose-6-phosphate isomerase deficiency affects step 2 of glycolysis. Triosephosphate isomerase deficiency affects step 5 of glycolysis. Phosphoglycerate kinase deficiency affects step 7 of glycolysis. Pyruvate kinase deficiency affects the 10th and last step of glycolysis.
Glucose-6-phosphate dehydrogenase deficiency affects the degradation of glucose in the pentose phosphate pathway, which is especially important in red blood cells.
For further information on inborn errors of glucose metabolism and inborn errors of glycogen metabolism see below.
Lactose
Lactose is a disaccharide sugar composed of galactose and glucose that is found in milk. Lactose can not be absorbed by the intestine and needs to be split in the small intestine into galactose and glucose by the enzyme called lactase; unabsorbed lactose can cause abdominal pain, bloating, diarrhea, gas, and nausea.
In most mammals, production of lactase diminishes after infants are weaned from maternal milk. However, 5% to 90% of the human population possess an advantageous autosomal mutation in which lactase production persists after infancy. The geographic distribution of lactase persistence is concordant with areas of high milk intake. Lactase non-persistence is common in tropical and subtropical countries. Individuals with lactase non-persistency may experience nausea, bloating and diarrhea after ingesting dairy.
Galactose
Galactosemia, the inability to metabolize galactose in liver cells, is the most common monogenic disorder of carbohydrate metabolism, affecting 1 in every 55,000 newborns. When galactose in the body is not broken down, it accumulates in tissues. The most common signs are failure to thrive, hepatic insufficiency, cataracts and developmental delay. Long term disabilities include poor growth, mental retardation, and ovarian failure in females.[2]
Galactosemia is caused by mutations in the gene that makes the enzyme galactose-1-phosphate uridylyltransferase. Approximately 70% of galactosemia-causing alleles have a single missense mutation in exon 6. A milder form of galactosemia, called Galactokinase deficiency, is caused a lack of the enzyme uridine diphosphate galactose-4-epimerase which breaks down a byproduct of galactose. This type of is associated with cataracts, but does not cause growth failure, mental retardation, or hepatic disease. Dietary reduction of galactose is also the treatment but not as severe as in patients with classical galactosemia. This deficiency can be systemic or limited to red blood cells and leukocytes.
Screening is performed by measuring GAL-1-P urydil transferase activity. Early identification affords prompt treatment, which consists largely of eliminating dietary galactose.
Fructose
Fructose malabsorption is a digestive disorder in which absorption of fructose is impaired by deficient fructose carriers in the small intestine's enterocytes.
Three autosomal recessive disorders impair fructose metabolism in liver cells. The most common is caused by mutations in the gene encoding hepatic fructokinase, an enzyme that catalyzes the first step in the metabolism of dietary fructose. Inactivation of the hepatic fructokinase results in asymptomatic fructosuria.
Hereditary fructose intolerance (HFI) results in poor feeding, failure to thrive, chronic liver disease and chronic kidney disease, and death. HFI is caused by a deficiency of fructose 1,6-biphosphate aldolase in the liver, kidney cortex and small intestine. Infants and adults are asymptomatic unless they ingest fructose or sucrose.
Deficiency of hepatic fructose 1,6-biphosphate (FBPase) causes impaired gluconeogenesis, hypoglycemia and severe metabolic acidemia. If patients are adequately supported beyond childhood, growth and development appear to be normal.
Essential fructosuria is a clinically benign condition characterized by the incomplete metabolism of fructose in the liver, leading to its excretion in urine.
By affected system
Glucose metabolism
Glycolysis
The metabolic pathway glycolysis is used by cells to break down carbohydrates like glucose (and various other simple sugars) in order to extract energy from them. During glycolysis ATP, NADH (both an energy transport form used inside cells) as well as pyruvate are produced.
Glycolysis is taking place in the cytosol where, under anaerobic conditions, pyruvate is converted to lactate. Under aerobic conditions, the pyruvate is transported from the cytosol to the mitochondrion, where further energy can be extracted through the citric acid cycle (CAC) (see below, c.f. bioenergetic systems).
The liver can also create glucose (gluconeogenesis, see below); during times of low carbohydrate supply from the digestive system, the liver creates glucose and supplies it to other organs.[3] Most enzymes of glycolysis also participate in gluconeogenesis, as it is mostly the reverse metabolic pathway of glycolysis; a deficiency of these liver enzymes will therefore impact both glycolysis and gluconeogenesis. (Note: gluconeogenesis is taking place only in the liver and not in other cells like e.g. muscle cells.)
| Glycolytic step Enzyme | Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Management and treatment | References and links |
|---|---|---|---|---|---|
| Glycolysis step 1 Glucokinase | GCK: Pancreatic beta cells Hyperinsulinemic hypoglycemia, familial, 3 (HHF3, hyperinsulinism due to glucokinase deficiency) | Hypoglycemia due to hyperinsulinemia. | NLM/GHR:GCK OMIM:GCK OMIM:HHF3 GARD:HHF3 ORPHA:HHF3 | ||
| Glycolysis step 1 Glucokinase | GCK: Pancreatic beta cells Maturity onset diabetes of the young type II (MODY2, GCK-MODY) | Diabetes. Hyperglycemia due to hypoinsulinemia while fasting but some glucose tolerance when consuming carbohydrates. | NLM/GHR:GCK OMIM:GCK OMIM:MODY2 GARD:MODY2 ORPHA:MODY2 | ||
| Glycolysis step 2 Glucose-6-phosphate isomerase | GPI: RBCs Glucose-6-phosphate isomerase deficiency (GPI deficiency, GPID, hemolytic anemia due to glucophosphate isomerase deficiency) | Hemolytic anemia. | NLM/GHR:GPI OMIM:GPI NLM/GHR:GPID OMIM:GPID ORPHA:GPID | ||
| Glycolysis step 3 Phosphofructo-kinase 1 (Not involved in glyconeogenesis) | PFKM: Muscle, also RBCs PFKL: Liver, also RBCs GSD type VII (GSD 7, Tarui's Disease, Phosphofructokinase deficiency) | Classic form: Symptoms usually appear in early childhood. Myopathy. Exercise-induced muscle cramps, weakness and sometimes rhabdomyolysis. Nausea and vomiting following strenuous exercise. Myoglobinuria, haemolytic anaemia, Hyperuricemia is common. High levels of bilirubin and jaundiced appearance possible. Late-onset form: Presents later in life. Myopathy, weakness and fatigue. Exercise intolerance (more than in GSD 5). Severe symptoms from classic type are absent. Infantile form: Rare. Often floppy infant syndrome (hypotonia), arthrogryposis, encephalopathy, cardiomyopathy and respiratory issues. Also central nervous system manifest possible, usually seizures. Hemolytic form: The defining characteristic is hemolytic anemia. Myopathy not as common. Rhabdomyolysis/myoglobinuria may cause acute renal failure. | Exercise test: Late about 3 times increase of lactate (higher than in GSD 5 and lower than in healthy). Increased rise of ammonia.[4] | No specific treatment. General advice is avoidance of vigorous exercise and of high-carbohydrate meals. | NLM/GHR:PFKM OMIM:PFKM OMIM:PFKL NLM/GHR:GSD VII OMIM:GSD VII GARD:GSD VII ORPHA:GSD VII |
| Glycolysis step 4 Aldolase A | ALDOA: Muscle, also liver and RBCs GSD type XII (GSD 12, Aldolase A deficiency, ALDOA deficiency, red cell aldolase deficiency) | Muscle Symptoms: Myopathy. Exercise intolerance, cramps. In some rhabdomyolysis and myoglobinuria. Liver Symptoms: In some Hepatomegaly RBC Symptoms: Hemolytic anemia. Rhabdomyolysis/myoglobinuria may cause acute renal failure. | Exercise test: ? | No treatment information in references given. | NLM/GHR:ALDOA OMIM:ALDOA OMIM:GSD XII GARD:GSD XII ORPHA:GSD XII |
| Glycolysis step 4 Aldolase B | ALDOB: Liver Hereditary fructose intolerance (Aldolase B deficiency, ALDOB deficiency) | Hypoglycemia. Hepatic and renal dysfunction. | NLM/GHR:ALDOB OMIM:ALDOB NLM/GHR:ALDOB D OMIM:ALDOB D GARD:ALDOB D ORPHA:ALDOB D | ||
| Glycolysis step 4 Aldolase C | ALDOC: Brain Unclear role in: | Neurodegeneration, unclear role. | See respective conditions | See respective conditions | OMIM:ALDOC |
| Glycolysis step 5 Triosephosphate isomerase | TPI1: RBCs Triosephosphate isomerase deficiency (TPID) | Hemolytic anemia. Reticulocytosis and hyperbilirubinemia are common. Classical generalized form: Progressive neurologic dysfunction with dystonia, tremor, dyskinesia, pyramidal tract signs, cardiomyopathy and spinal motor neuron involvement with progressive neuromuscular impairment (severe weakness and muscle wasting). | NLM/GHR:TPI1 OMIM:TPI1 NLM/GHR:TPID OMIM:TPID GARD:TPID ORPHA:TPID | ||
| Glycolysis step 6 Glyceraldehyde 3-phosphate dehydrogenase | GAPDH: Brain Unclear role in: | Neurodegeneration, unclear role. | See respective conditions | See respective conditions | OMIM:GAPDH |
| Glycolysis step 7 Phosphoglycerate kinase | PGK1: Muscle, RBCs Phosphoglycerate kinase deficiency (PGK1D, PGK deficiency, GSD due to phosphoglycerate kinase 1 deficiency) | Myopathic form: Progressive muscle weakness, pain, and cramping, particularly with exercise. Myoglobinuria possible. Myoglobinuria may cause acute renal failure. Hemolytic form: Hemolytic anemia. Neurologic form: In some central nervous system manifestation, including hemiplegic migraines, epilepsy, ataxia and tremor. Progressive neurologic impairment in some. Combinations of 1, 2 or all 3 forms have been reported. | Exercise test: ? | Regular blood transfusions for severe chronic anemia; splenectomy has been shown to be beneficial in some cases. | NLM/GHR:PGK1 OMIM:PGK1 NLM/GHR:PGK1D OMIM:PGK1D GARD:PGK1D ORPHA:PGK1D |
| Glycolysis step 8 Phosphoglycerate mutase | PGAM2: Muscle GSD type X (GSD 10, muscle phosphoglycerate mutase deficiency, myopathy due to PGAM deficiency, PGAMD) | Myopathy, exercise intolerance. Exercise-induced cramps, myoglobinuria and myalgia. Rhabdomyolysis possible. Rhabdomyolysis/myoglobinuria may cause acute renal failure. | Exercise test: ? | No treatment information in references given. | NLM/GHR:PGAM2 OMIM:PGAM2 NLM/GHR:GSD X OMIM:GSD X GARD:GSD X ORPHA:GSD X |
| Glycolysis step 9 Enolase 1 (Alpha-enolase, α-enolase) | ENO1: RBCs Enolase deficiency (α-enolase deficiency, alpha-enolase deficiency) | Hemolytic anemia. | OMIM:ENO1 | ||
| Glycolysis step 9 Enolase 1 (Alpha-enolase, α-enolase) | ENO1 Unclear role in: | Autoimmunity, unclear role. | See respective conditions | See respective conditions | OMIM:ENO1 |
| Glycolysis step 9 Enolase 3 (Beta-enolase, β-enolase) | ENO3: Muscle GSD type XIII (GSD 13, β-enolase deficiency, beta-enolase deficiency, enolase 3 deficiency, muscle enolase deficiency) | Myopathy. Exercise-induced myalgias, generalized muscle weakness and fatigability. | Exercise test: No rise of lactate. Biopsy: Focal sarcoplasmic accumulation of glycogen-beta particles. Immunohistochemistry and immunoblotting show reduced beta-enolase protein. | No treatment information in references given. | NLM/GHR:ENO3 OMIM:ENO3 OMIM:GSD XIII GARD:GSD XIII ORPHA:GSD XIII |
| Glycolysis step 10 Pyruvate kinase | PKLR: RBCs, liver Pyruvate kinase deficiency (PK deficiency, PKD) | Hemolytic anemia. | NLM/GHR:PKLR OMIM:PKLR NLM/GHR:PKD OMIM:PKD GARD:PKD ORPHA:PKD |
Related to glycolysis
The pyruvate created by glycolysis (in the cytosol) is transported (together with a proton) to the mitochondrion for further energy extraction.
Under anaerobic conditions (without the use of oxygen) most if not all of the pyruvate is converted into lactate (furthermore producing NAD+ at the same time).
Under aerobic conditions (with the use of oxygen) only part of the pyruvate is converted to lactate; the pyruvate not converted feeds the citric acid cycle (CAC); both via pyruvate dehydrogenase (PDC, with Acetyl-CoA as intermediate) and via pyruvate decarboxylation - this will create further ATP and NADH for the cell's use.
The pentose phosphate pathway (HMP Shunt) is connected to the glycolysis pathway, and can convert substrates to and from the glycolysis pathway. It generates NADPH and pentoses (5-carbon sugars) as well as ribose 5-phosphate, a precursor for the synthesis of nucleotides. While the pentose phosphate pathway does involve oxidation of glucose, its primary role is anabolic rather than catabolic. The pathway is especially important in red blood cells (erythrocytes).
Transport proteins move substrates through cellular membranes. A glucose transporter (GLUT) protein is needed to assist glucose into (and in the liver and kidneys, also out of) the cell. De Vivo disease (GLUT1 deficiency) is a deficiency of GLUT1, which is needed to transport glucose across the blood-brain barrier. Fanconi-Bickel syndrome (GLUT2 deficiency, formally known as GSD-XI) is a deficiency of GLUT2, which is needed for the transport of glucose between liver and blood.
Mitochondrial pyruvate carrier deficiency (MPYCD) is a metabolic disorder, in which the transport of pyruvate from the cytosol to the mitochondria is affected (gene SLC54A1/BRP44L/MPC1[5]); the deficiency is characterized by delayed psychomotor development and lactic acidosis with a normal lactate/pyruvate ratio resulting from impaired mitochondrial pyruvate oxidation.[6] A similar disease is also seen in mutations of gene SLC54A2/BRP44/MPC2.[7]
The gene SLC16A1/MCT1 is responsible for transporting lactate across membranes. Mutations in the monocarboxylate transporter 1 (MCT1) gene have been associated with three diseases: hyperinsulinemic hypoglycemia, familial 7 (HHF7); monocarboxylate transporter 1 deficiency (MCTD1); and erythrocyte lactate transporter defect (formerly, myopathy due to lactate transport defect).[8]
(See also bioenergetic systems.)
| Related enzymatic function – Enzyme (Relation) | Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Management and treatment | References and links |
|---|---|---|---|---|---|
| Pyruvate dehydrogenation / decarboxylation. – Pyruvate dehydrogenase complex
| PDHA1, DLD, PDHX, PDHB, DLAT, PDP1, LIAS Systemic/various Pyruvate dehydrogenase deficiency (PDHA deficiency, PDHAD, ataxia with lactic acidosis, intermittent ataxia with pyruvate dehydrogenase deficiency, pyruvate dehydrogenase complex deficiency, pyruvate decarboxylase deficiency, pyruvate dehydrogenase lipoic acid synthetase deficiency (PDHLD), some forms of Leigh Syndrome (genes PDHA1 and DLD)) | 2 major presentations: metabolic and neurologic. Between these 2 major presentations, there is a continuous spectrum of intermediate forms.[9] Those with predominantly neurological symptoms fit within the category of Leigh Syndrome.[9]
Broad clinical spectrum: From fatal lactic acidosis in the newborn to chronic neurologic dysfunction with structural abnormalities in the central nervous system without systemic acidosis. Most common cause of primary lactic acidosis in children. | NLM/GHR:PDHA1 OMIM: PDHA1 OMIM: DLD OMIM: PDHX OMIM: PDHB OMIM: DLAT OMIM: PDP1 OMIM: LIASNLM/GHR:PDHAD OMIM: Leigh Syndrome OMIM: PDHAD
OMIM: DLDD
OMIM: PDHXD
OMIM: PDHBD OMIM: PDHPD OMIM: PDHLD | ||
| Inter-conversion of pyruvate and lactate. – Lactate dehydrogenase A
| LDHA: Muscle GSD type XI (GSD 11, lactate dehydrogenase deficiency, LDH deficiency) | Myopathy. Exercise intolerance.
Note: Deficiency of dehydrogenase-B (LDHB) has been observed as asymptomatic. | Exercise test: Increased pyruvate, but no rise of lactate. | No treatment information in references given. | NLM/GHR:LDHA OMIM: LDHA NLM/GHR: GSD 11 OMIM: GSD 11 ORPHA: GSD 11 |
| Inter-conversion between the pentose phosphate pathway and the glycolysis pathway (Fructose-6-P, Glyceraldehyde-3-P, Erythrose-4-P, Xylulose-5-P, and Ribose-5-P) – Transketolase |
TKT: multi-organ Transketolase Deficiency (SDDHD [Short Stature, Developmental Delay, and congenital Heart Defects]) |
Developmental delay and intellectual disability, delayed or absent speech, short stature, and congenital heart defects. Additional features reported. | Elevated plasma and urinary polyols (erythritol, arabitol, and ribitol) and urinary sugar-phosphates (ribose-5-phosphate and xylulose/ribulose-5-phosphate). Biopsy shows absent or low TKT. | OMIM: SDDHD
ORPHA: TKT |
Gluconeogenesis
| Gluconeogenesis step – Enzyme
| Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Treatment | References and links |
|---|---|---|---|---|---|
| Gluconeogenesis step 1:
Conversion of pyruvate into oxaloacetate – Pyruvate carboxylase |
PC: Liver Pyruvate carboxylase deficiency types A, B, & C (Leigh necrotizing encephalopathy due to pyruvate carboxylase deficiency, Ataxia with lactic acidosis II) |
Lactic acidosis, hyperammonemia, hypoglycemia, hepatomegaly, neurological problems, hypotonia, reduced ketone synthesis, and demyelination of neurons. | Some may respond to thiamine treatment | OMIM: PC OMIM: PC deficiency | |
| Gluconeogenesis step 8 – Fructose 1,6-bisphosphatase
| FBP1: Liver Fructose bisphosphatase deficiency (FBP1, Baker-Winegrad disease) | Fasting hypoglycemia with lactic acidosis. Episodes of hyperventilation, apnea, and ketosis. Symptoms exacerbated by fructose, sucrose, and glycerol consumption. | NLM/GHR:FBP1 OMIM:FBP1 OMIM:FBP1D GARD:FBP1D ORPHA:FBP1D | ||
| Gluconeogenesis step 10 (final step): Conversion of G-1-P into glucose – Glucose 6-phosphatase
| G6PC: Liver SLC37A4 (G6PT1): Liver GSD type I (GSD 1, von Gierke's disease, hepatorenal glycogenosis, glucose-6-phosphate deficiency, glucose-6-phosphate transport defect) | Hypoglycemia and hepatomegaly. Growth retardation, delayed puberty, lactic acidemia, hyperlipidemia, hyperuricemia. In adults hepatic adenomas likely. | Exercise test: Normal lactate and ammonia rise.[10] | NLM/GHR:G6PC OMIM:G6PC NLM/GHR:SLC37A4 OMIM:SLC37A4 NLM/GHR:GSD 1 ORPHA:GSD 1 OMIM:GSD 1a GARD:GSD 1a ORPHA:GSD 1a OMIM:GSD 1b GARD:GSD 1b ORPHA:GSD 1b OMIM:GSD 1c/1d | |
| Gluconeogenesis step 10 (final step): Conversion of G-1-P into glucose – Glucose 6-phosphatase
| G6PC3: WBCs, heart, others Severe congenital neutropenia type 4 (SCN4, congenital agranulocytosis, congenital neutropenia, Kostmann's disease, severe congenital neutropenia-pulmonary hypertension-superficial venous angiectasis) Dursun syndrome (DURSS, pulmonary arterial hypertension-leukopenia-atrial septal defect syndrome) | SCN4: A disorder of hematopoiesis. Maturation arrest of granulopoiesis at the level of promyelocytes. Neutropenia. Osteopenia, may lead to osteoporosis. Prone to recurrent infections. In some heart and genital abnormalities, cancerous conditions of the blood, seizures, developmental delay. Dursun syndrome: Pulmonary arterial hypertension, cardiac abnormalities (including secundum-type atrial septal defect), intermittent neutropenia, lymphopenia, monocytosis and anemia. | NLM/GHR:G6PC3 OMIM:G6PC3 NLM/GHR:SCN4 OMIM:SCN4 ORPHA:SCN4 ORPHA:DURSS |
Glycogen metabolism
Glycogenesis
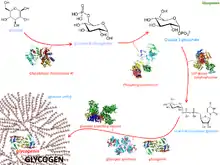
Glycogenesis is the metabolic pathway in which glycogen is created. Glycogen, which consists of branched long chains made out of the simple sugar glucose, is an energy storage form for carbohydrates in many human cells; this is most important in liver, muscle and certain brain cells.
The monosaccharide glucose-6-phosphate (G-6-P) is typically the input substance for glycogenesis. G-6-P is most commonly created from glucose by the action of the enzymes glucokinase (see glycolysis step 1) or hexokinase.
Through the action of several enzymes glycogen is built up:
- G-6-P is converted into glucose-1-phosphate (G-1-P) by the action of phosphoglucomutase (PGM), passing through the obligatory intermediate glucose-1,6-bisphosphate.
- G-1-P is converted into UDP-glucose by the action of the enzyme UDP-glucose pyrophosphorylase (UGP).
- The enzyme glycogenin (GYG) is needed to create initial short glycogen chains, which are lengthened and branched by the other enzymes of glycogenesis.
- Once eight glucose have been added to the glycogen chain, then glycogen synthase (GYS) can bind to the growing glycogen chain and add UDP-glucose to lengthen the glucogen chain.
- Branches are made by glycogen branching enzyme (GBE), which transfers the end of the chain onto an earlier part, forming branches; these grow further grow by addition of more units.
On an alternative metabolic pathway the simple sugar galactose (Gal, which is typically derived from lactose) is converted by the enzyme galactokinase (GALK) to galactose-1-phosphate (Gal-1-P), which in turn is converted by the enzyme galactose-1-phosphate uridylyltransferase (GALT) to glucose-1-phosphate (G-1-P), which can also serve as input for glycogenesis – this bypasses the first step of glycogenesis (the enzyme phosphoglucomutase PGM).
Errors in glycogenesis can have different consequences on a cellular level:
- Too little glycogen is produced, e.g. in GSD 0
- The glycogen is badly formed and inaccessible, typically accumulating in the affected cells (e.g. polyglucosan bodies).
Depending on the affected cells and the extent of the deficiency, a wide range of symptoms and severities are the result.
| Glycogenesis step – Enzyme | Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Treatment | References and links |
|---|---|---|---|---|---|
| Glycogenesis step: Inter-conversion of G-1-P and G-6-P – Phosphoglucomutase 1 (Also last step of glycogenolysis) | PGM1: Liver, muscle, other CDG syndrome type It (CDG1T, PGM1-CDG, phosphoglucomutase 1 deficiency, PGM1 deficiency) formerly: GSD type XIV (GSD 14) | Wide range of manifestations and severity. Commonly cleft lip and bifid uvula, hepatopathy, intermittent hypoglycemia, short stature, and exercise intolerance. | DNA Test: mutation on PGM1.
Walk test: Second Wind phenomenon in some,[11] but not all.[12] Observable with treadmill and heart rate monitor. Muscle biopsy: shows glycogen accumulation. Blood test: Abnormal serum transferrin. Non-ischemic forearm test: exercise-induced hyperammonemia with normal lactic acid rise.[13] | NLM/GHR:PGM1 OMIM:PGM1 OMIM:CDG 1T ORPHA:CDG 1T | |
| Glycogenesis step: UDP-glucose synthesis – UDP-glucose pyrophosphorylase
| UGP2 Barakat-Perenthaler syndrome, EPILEPTIC ENCEPHALOPATHY, EARLY INFANTILE, 83; EIEE83 | severe autosomal recessive neurodevelopmental disorder, presenting in early life with intractable seizures, absence of virtually all developmental milestones, visual impairment, progressive microcephaly and minor dysmorphic features[14] | - | - | OMIM:UGP2 |
| Glycogenesis step: Glycogen primer synthesis – Glycogenin
| GYG1: Muscle GSD type XV (GSD 15, glycogenin deficiency) Polyglucosan body myopathy type 2 (PGBM2) | GSD 15: Myopathy, cardiomyopathy. Rare. Muscle weakness. PGBM2: Myopathy. Proximal muscle weakness of the lower limbs, gait disturbances. Upper limbs and/or distal muscle weakness in some. Onset-age highly variable, slowly progressive. | Exercise test: ? Skeletal muscle biopsy: deficit of glycogen, predominance of slow-twitch, oxidative muscle fibers and mitochondrial proliferation. Endomyocardial biopsy: hypertrophic cardiomyocytes, enlarged nuclei and large centrally located vacuoles containing periodic acid Schiff (PAS)-positive material (but ultrastructurally different from glycogen). Glycogen depletion in the remainder of the cytoplasm. | No treatment information in references given. | NLM/GHR:GYG1 OMIM:GYG1 OMIM:GSD 15 ORPHA:GSD 15 |
| Glycogenesis step: Glucogen chain lengthening – Glycogen synthase
| GYS1: Muscle GSD type 0b (GSD 0b, glycogen synthetase deficiency) | Myopathy, cardiomyopathy, exercise intolerance. | Exercise test: ? | NLM/GHR:GYS2 OMIM:GYS2 OMIM:GSD 0B ORPHA:GSD 0B | |
| Glycogenesis step: Glucogen chain lengthening – Glycogen synthase
| GYS2: Liver GSD type 0a (GSD 0a, glycogen synthetase deficiency) | Infancy or in early childhood onset. Morning fatigue and fasting hypoglycemia, hyperketonemia. Without hepatomegaly, hyperalaninemia or hyperlactacidemia. After meals, major hyperglycemia associated with lactate and alanine increase and hyperlipidemia. | NLM/GHR:GYS2 OMIM:GYS2 NLM/GHR:GSD 0 OMIM:GSD 0A ORPHA:GSD 0A | ||
| Glycogenesis step: Glucogen chain branching – Glycogen branching enzyme
| GBE1: Liver, muscle GSD type IV (GSD 4, Andersen's disease, amylopectinosis, brancher deficiency, glycogen branching enzyme deficiency, familial cirrhosis with deposition of abnormal glycogen) | Different forms have been described: | Activity of branching enzyme in erythrocytes. | High-protein diet. Liver transplant for progressive liver disease. Cardiomyopathy may require certain medications. | NLM/GHR:GBE1 OMIM:GBE1 NLM/GHR:GSD 4 OMIM:GSD 4 GARD:GSD 4 ORPHA:GSD 4 |
| Glycogenesis step: Glucogen chain branching – Glycogen branching enzyme
| GBE1: Nerve cells Adult polyglucosan body disease (APBD) | Neuropathy, affecting the central and peripheral nervous systems. Cognitive impairment, pyramidal tetraparesis, peripheral neuropathy, and neurogenic bladder. Peripheral neuropathy and progressive muscle weakness and stiffness (spasticity). Cerebellar dysfunction and extrapyramidal signs in some. Late-onset, slowly progressive. | NLM/GHR:GBE1 OMIM:GBE1 NLM/GHR:APBD OMIM:APBD GARD:APBD ORPHA:APBD |
Glycogenolysis

To access the energy stored as glycogen, cells use the metabolic pathway glycogenolysis (glycogen breakdown); this produces the simple sugar glucose-6-phosphate (G-6-P), from which cells can extract energy or build other substances (e.g. riboses).
G-6-P (which is also produced from glucose) acts as an input substance for:
- Glycolysis (see above)
- The Pentose phosphate pathway (PPP)
(See also bioenergetic systems.)
An alternative to glycolysis is the Pentose phosphate pathway (PPP): Depending on cellular conditions the PPP can produce NADPH (another energy transport form in the cell) or synthesize riboses (important for substances based on ribose like e.g. RNA) - the PPP is for example important in red blood cells.
If glycogenolysis is taking place in the liver, G-6-P can be converted to glucose by the enzyme glucose 6-phosphatase (G6Pase); the glucose produced in the liver is then released to the bloodstream for use in other organs. Muscle cells in contrast do not have the enzyme glucose 6-phosphatase, so they cannot share their glycogen stores with the rest of the body.
In addition to glycogen breakdown with the glycogen debranching enzyme and the glycogen phosphorylase enzyme, cells also use the enzyme acid alpha-glucosidase in lysosomes to degrade glycogen.
A deficiency of an involved enzyme results in:
- Accumulation of glycogen in the cells
- Lack of cellular energy negatively affects the involved organs
Myophosphorylase (muscle glycogen phosphorylase) comes in two forms: form 'a' is phosphorylated by phosphorylase kinase, form 'b' is not phosphorylated. Form 'a' is de-phosphorylated into form 'b' by the enzyme phosphoprotein phosphatase, which is activated by elevated insulin.
Both forms 'a' and 'b' of myophosphorylase have two conformational states: active (R or relaxed) and inactive (T or tense). When either form 'a' or 'b' are in the active state, then the enzyme converts glycogen into glucose-1-phosphate.
Myophosphorylase-b is allosterically activated by elevated AMP within the cell, and allosterically inactivated by elevated ATP and/or glucose-6-phosphate. Myophosphorylase-a is active, unless allosterically inactivated by elevated glucose within the cell. In this way, myophosphorylase-a is the more active of the two forms as it will continue to convert glycogen into glucose-1-phosphate even with high levels of glycogen-6-phosphate and ATP. (See Glycogen phosphorylase§Regulation).
| Glycogenolysis step – Enzyme | Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Treatment | References and links |
|---|---|---|---|---|---|
| Glycogenolysis step: Release of G-1-P – Glycogen phosphorylase
| PYGL: Liver GSD type VI (GSD 6, Hers disease, hepatic glycogen phosphorylase deficiency, liver phosphorylase deficiency syndrome) | Hepatomegaly, failure to thrive, growth retardation. No other developmental delay, no muscle involvement. Hypoglycemia, lactic acidosis, hyperlipidemia and ketosis during prolonged fasting periods. Infancy or childhood onset, symptoms tend to improve with age. | NLM/GHR:PYGL OMIM:PYGL NLM/GHR:GSD 6 OMIM:GSD 6 GARD:GSD 6 ORPHA:GSD 6 | ||
| Glycogenolysis step: Release of G-1-P – Glycogen phosphorylase
| PYGM: Muscle GSD type V (GSD 5, McArdle's disease, muscle phosphorylase deficiency, myophosphorylase deficiency, PYGM deficiency) | Myopathy: Exercise intolerance, symptoms tend to improve with rest. "Second wind" phenomenon in most. Some have hypertrophic calf muscles.[15] Rhabdomyolysis and myoglobinuria possible. Some have muscle weakness. Of those with muscle weakness, in two-thirds it worsens, however in some the muscle weakness is stable. Onset forms: infant, child, adult. Infant-form most severe (e.g. progressive respiratory failure), adult-onset can be very mild (e.g. mainly poor stamina). | Exercise test: Severely impaired rise of lactate. Normal or enhanced ammonia.[10][4][16] Exercise-induced inappropriate rapid heart rate and myogenic hyperuricemia.[17][18] "Second wind" observable with treadmill and heart rate monitor in a 12-minute walk test.[19][20]
Muscle biopsy: Skeletal muscle shows abnormal glycogen accumulation.[17] DNA test: Autosomal recessive mutation on PYGM gene.[17] | NLM/GHR:PYGM OMIM:PYGM NLM/GHR:GSD 5 OMIM:GSD 5 GARD:GSD 5 ORPHA:GSD 5 | |
| Glycogenolysis step: Release of G-1-P – Glycogen phosphorylase |
PYGM: Muscle Myophosphorylase-a activity impaired, myophosphorylase-b preserved |
Adult-onset muscle weakness. No exercise intolerance (which differentiates it from McArdle disease, GSD-V) | DNA test: Autosomal dominant mutation on PYGM gene.
Muscle biopsy: Accumulation of the intermediate filament desmin in the myofibers |
PMID: 32386344 | |
| Glycogenolysis step: Debranching of PLD – Glycogen debranching enzyme (GDE)
| AGL: Liver, muscles GSD type III (GSD 3, Forbes disease, Cori disease, limit dextrinosis, glycogen debrancher deficiency, GDE deficiency, AGL deficiency) | Infant or child onset, often at puberty some symptoms improve. Liver: Hepatomegaly, growth retardation, hyperlipidemia, hypoglycemia. Occasional seizures related to hypoglycemia. Adult cirrhosis in some. Muscle: Myopathy, muscular hypotonia, muscle wasting (distal, some limb-girdle, some proximal instead), hypertrophic cardiomyopathy. Slowly progressive muscle weakness. GSD IIIa / IIIc: Liver and muscle GSD IIIb / IIId: Liver only | Exercise test: Severely impaired lactate response (muscle involvement). Normal or enhanced ammonia.[10] | NLM/GHR:AGL OMIM:AGL NLM/GHR:GSD 3 OMIM:GSD 3 GARD:GSD 3 ORPHA:GSD 3 |
Related to glycogenolysis
| Affected enzymatic function – Enzyme (Relation to glycogenolysis) | Gene: Organ(s) Disease (Synonyms) | Reported symptoms. Forms (if applicable) Note: Not all patients have all symptoms; severity and presentation can vary. | Diagnostic tests | Treatment | References and links |
|---|---|---|---|---|---|
| Glycogenolysis final step: Release of G-1-P – Phosphorylase kinase, alpha 1 (Activation of liver glycogen phosphorylase, c.f. GSD 6) | PHKA2: Liver (GSD 9a) PHKB: Liver, Muscle (GSD 9b) PHKG2: Liver (GSD 9c) GSD type IX | GSD 9a: Liver form. Hepatomegaly, growth retardation, elevation of glutamate-pyruvate transaminase and glutamate-oxaloacetate transaminase, hypercholesterolemia, hypertriglyceridemia, and fasting hyperketosis. Improves with age, most adult patients are asymptomatic. GSD 9a1: PhK deficiency in erythrocytes. GSD 9a2: Normal PhK activity in erythrocytes. GSD 9b: Liver and muscle form. Additionally mild myopathy like GSD 9d. Rare. GSD 9c: Similar to GSD 9a, but tends to be more severe. In some hepatic fibrosis or cirrhosis. | Exercise test: Muscle involvement see GSD 9d. | NLM/GHR:PHKA2 OMIM:PHKA2 NLM/GHR:PHKB OMIM:PHKB NLM/GHR:PHKG2 OMIM:PHKG2 NLM/GHR:GSD 9 ORPHA:GSD 9 OMIM:GSD 9a1/9a2 ORPHA:GSD 9a/9c OMIM:GSD 9b ORPHA:GSD 9b OMIM:GSD 9c | |
| Glycogenolysis final step: Release of G-1-P – Phosphorylase kinase, alpha 1 (Activation of muscle glycogen phosphorylase, c.f. GSD 5) | PHKA1: Muscle GSD type IXd (GSD 9d, phosphorylase b kinase deficiency, PhK deficiency, muscle glycogenosis) Formerly GSD type VIII (GSD 8) Formerly GSD type Vb (GSD 5b)[21] | Myopathy. Exercise-induced muscle weakness or stiffness. Relative mild compared to other metabolic myopathies. Typically adult-onset, some asymptomatic in late adulthood. No liver involvement. | Exercise test: Both impaired and normal lactate observed; possible submaximal/maximal or aerobic/anaerobic discrepancy. Normal or exaggerated ammonia response.[22] | NLM/GHR:PHKA1 OMIM:PHKA1 NLM/GHR:GSD 9 OMIM:GSD 9d ORPHA:GSD 9d/9e | |
| Degradation of glycogen to glucose in lysosomes – Acid alpha-glucosidase
Lysosome-associated membrane protein 2
| GAA: Myopathy Myopathy GSD type II (GSD 2a, Pompe's disease, acid maltase deficiency, deficiency of lysosomal alpha-glucosidase, cardiomegalia glycogenica) Danon disease (GSD 2b, Danon disease, lysosomal glycogen storage disease without acid maltase deficiency) | Symptoms of both GSD types IIa and IIb are very similar due to a defect in lysosomes. However, in type IIb, some show abnormal glycogen accumulation, but not all.
Classic infantile form (Pompe disease): Cardiomyopathy and muscular hypotonia. In some respiratory involvement. | DNA test: Mutation in either GAA or LAMP2 gene. Pompe disease is autosomal recessive. Danon disease is X-linked dominant.
Muscle biopsy: Abnormal glycogen accumulation in lysosomes. | NLM/GHR:GAA OMIM:GAAOMIM: LAMP2 NLM/GHR:GSD 2 OMIM:GSD 2 GARD:GSD 2 ORPHA:GSD 2OMIM: Danon disease |
Mutations in the PRKAG2 gene have been traced to fatal congenital nonlysosomal cardiac glycogenosis; PRKAG2 is a noncatalytic gamma subunit of AMP-activated protein kinase (AMPK), which affects the release of G-1-P by phosphorylase kinase during nonlysosomal glycogenolysis.[23]
See also
- Glycogen Storage Disease
- Metabolic Myopathies
- Exercise intolerance § low ATP reservoir
- Myogenic hyperuricemia
- Purine nucleotide cycle § pathology (low ATP reservoir, ADP>ATP, ↑AMP)
- Tachycardia § sinus (inappropriate rapid heart rate response to exercise)
- IST § differential diagnoses
- Second Wind (exercise phenomenon)
References
- ↑ Jorde, et al. 2006. Carbohydrate metabolism. Medical Genetics. 3rd edition. Chapter 7. Biochemical genetics:Disorders of metabolism. pp139-142.
- ↑ Suchy FJ, Sokol RJ, Balistreri WF (2007), Liver disease in children, Cambridge University Press, p. 598, ISBN 9781139464031
- ↑ Webster CC, Noakes TD, Chacko SK, Swart J, Kohn TA, Smith JA (August 2016). "Gluconeogenesis during endurance exercise in cyclists habituated to a long-term low carbohydrate high-fat diet". The Journal of Physiology. 594 (15): 4389–4405. doi:10.1113/JP271934. PMC 4967730. PMID 26918583.
- 1 2 Piirilä P, Similä ME, Palmio J, Wuorimaa T, Ylikallio E, Sandell S, et al. (2016). "Unique Exercise Lactate Profile in Muscle Phosphofructokinase Deficiency (Tarui Disease); Difference Compared with McArdle Disease". Frontiers in Neurology. 7: 82. doi:10.3389/fneur.2016.00082. PMC 4885106. PMID 27303362.
- ↑ OMIM:BRP44L
- ↑ OMIM:MPYCD
- ↑ Pujol C, Lebigot E, Gaignard P, Galai S, Kraoua I, Bault JP, et al. (March 2023). "MPC2 variants disrupt mitochondrial pyruvate metabolism and cause an early-onset mitochondriopathy". Brain. 146 (3): 858–864. doi:10.1093/brain/awac444. PMC 9976959. PMID 36417180.
- ↑ "SOLUTE CARRIER FAMILY 16 (MONOCARBOXYLIC ACID TRANSPORTER), MEMBER 1; SLC16A1". www.omim.org. Retrieved 2023-08-22.
- 1 2 Brown GK, Otero LJ, LeGris M, Brown RM (November 1994). "Pyruvate dehydrogenase deficiency". Journal of Medical Genetics. 31 (11): 875–879. doi:10.1136/jmg.31.11.875. PMC 1016663. PMID 7853374.
- 1 2 3 Livingstone C, Chinnery PF, Turnbull DM (July 2001). "The ischaemic lactate-ammonia test". Annals of Clinical Biochemistry. 38 (Pt 4): 304–310. doi:10.1258/0004563011900786. PMID 11471870. S2CID 23496022.
- ↑ Preisler N, Cohen J, Vissing CR, Madsen KL, Heinicke K, Sharp LJ, et al. (November 2017). "Impaired glycogen breakdown and synthesis in phosphoglucomutase 1 deficiency". Molecular Genetics and Metabolism. 122 (3): 117–121. doi:10.1016/j.ymgme.2017.08.007. PMID 28882528.
- ↑ Stojkovic T, Vissing J, Petit F, Piraud M, Orngreen MC, Andersen G, et al. (July 2009). "Muscle glycogenosis due to phosphoglucomutase 1 deficiency". The New England Journal of Medicine. 361 (4): 425–427. doi:10.1056/NEJMc0901158. PMID 19625727.
- ↑ Altassan R, Radenkovic S, Edmondson AC, Barone R, Brasil S, Cechova A, et al. (January 2021). "International consensus guidelines for phosphoglucomutase 1 deficiency (PGM1-CDG): Diagnosis, follow-up, and management". Journal of Inherited Metabolic Disease. 44 (1): 148–163. doi:10.1002/jimd.12286. PMC 7855268. PMID 32681750.
- ↑ Perenthaler, E., Nikoncuk, A., Yousefi, S. et al. Loss of UGP2 in brain leads to a severe epileptic encephalopathy, emphasizing that bi-allelic isoform-specific start-loss mutations of essential genes can cause genetic diseases. Acta Neuropathol 139, 415–442 (2020). https://doi.org/10.1007/s00401-019-02109-6
- ↑ Rodríguez-Gómez I, Santalla A, Díez-Bermejo J, Munguía-Izquierdo D, Alegre LM, Nogales-Gadea G, et al. (November 2018). "Non-osteogenic muscle hypertrophy in children with McArdle disease". Journal of Inherited Metabolic Disease. 41 (6): 1037–1042. doi:10.1007/s10545-018-0170-7. hdl:10578/19657. PMID 29594644. S2CID 4394513.
- ↑ Delaney NF, Sharma R, Tadvalkar L, Clish CB, Haller RG, Mootha VK (August 2017). "Metabolic profiles of exercise in patients with McArdle disease or mitochondrial myopathy". Proceedings of the National Academy of Sciences of the United States of America. 114 (31): 8402–8407. Bibcode:2017PNAS..114.8402D. doi:10.1073/pnas.1703338114. PMC 5547614. PMID 28716914.
- 1 2 3 Lucia A, Martinuzzi A, Nogales-Gadea G, Quinlivan R, Reason S (December 2021). "Clinical practice guidelines for glycogen storage disease V & VII (McArdle disease and Tarui disease) from an international study group". Neuromuscular Disorders. 31 (12): 1296–1310. doi:10.1016/j.nmd.2021.10.006. PMID 34848128. S2CID 240123241.
- ↑ Mineo I, Kono N, Hara N, Shimizu T, Yamada Y, Kawachi M, et al. (July 1987). "Myogenic hyperuricemia. A common pathophysiologic feature of glycogenosis types III, V, and VII". The New England Journal of Medicine. 317 (2): 75–80. doi:10.1056/NEJM198707093170203. PMID 3473284.
- ↑ Scalco RS, Chatfield S, Godfrey R, Pattni J, Ellerton C, Beggs A, et al. (July 2014). "From exercise intolerance to functional improvement: the second wind phenomenon in the identification of McArdle disease". Arquivos de Neuro-Psiquiatria. 72 (7): 538–541. doi:10.1590/0004-282x20140062. PMID 25054987.
- ↑ Pérez M, Ruiz JR, Fernández Del Valle M, Nogales-Gadea G, Andreu AL, Arenas J, Lucía A (June 2009). "The second wind phenomenon in very young McArdle's patients". Neuromuscular Disorders. 19 (6): 403–405. doi:10.1016/j.nmd.2009.04.010. PMID 19477644. S2CID 31541581.
- ↑ GeneReviews: Phosphorylase Kinase Deficiency
- ↑ OMIM:GSD 9d
- ↑ OMIM:PRKAG2
External links
 Media related to Disorders of carbohydrate metabolism at Wikimedia Commons
Media related to Disorders of carbohydrate metabolism at Wikimedia Commons