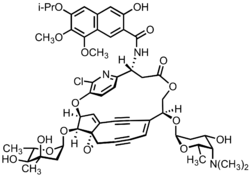
 | |
 | |
| Names | |
|---|---|
| IUPAC name
N-[(3S,9R,14S,15E,19S,21R,24R)-6-chloro-24-[(2S,4R,5S,6S)-4,5-dihydroxy-4,6-dimethyloxan-2-yl]oxy-14-[(2S,4S,5S,6S)-5-(dimethylamino)-4-hydroxy-6-methyloxan-2-yl]oxy-11-oxo-4,12,20-trioxa-7-azapentacyclo[13.6.2.25,8.13,21.019,21]hexacosa-1,5,7,15,25-pentaen-17,22-diyn-9-yl]-3-hydroxy-7,8-dimethoxy-6-propan-2-yloxynaphthalene-2-carboxamide | |
| Identifiers | |
| |
3D model (JSmol) |
|
| ChemSpider |
|
| KEGG |
|
PubChem CID |
|
| |
| |
| Properties | |
| C53H60ClN3O16 | |
| Molar mass | 1030.52 g·mol−1 |
| Appearance | Buff-colored amorphous solid |
| Hazards | |
| Occupational safety and health (OHS/OSH): | |
Main hazards |
Cytotoxic, mutagen |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
Kedarcidin is a chromoprotein antitumor antibiotic first isolated from an Actinomycete in 1992, comprising an ansa-bridged enediyne chromophore (shown) as well as an apoprotein that serves to stabilize the toxin in the Actinomycete. Like other members of the enediyne class of drugs—so named for the nine-or-ten-membered core structure bearing an alkene directly attached to two alkynyl appendages—kedarcidin was likely evolved to kill bacteria that compete with the producing organism. Because it achieves this by causing DNA damage, however, kedarcidin is capable of harming tumor cells, as well. Kedarcidin is thus the subject of scientific research, both for its structural complexity as well as its anticancer properties.
Discovery and structure elucidation
Kedarcidin was first discovered in 1992 when bioassays conducted at Bristol-Myers Squibb indicated the presence of a DNA-damaging chromoprotein in the fermentation broth of an Actinomycete strain. The involvement of a non-peptidic chromophore was deduced by UV spectroscopy, and reverse-phase chromatography was used to separate this noncovalently bound chromophore from its apoprotein host. This isolate—kedarcidin chromophore—decomposed readily under ambient conditions and was shown to possess cytotoxicity (IC50 0.4 ng/ml, HCT-116 human colorectal carcinoma cell line).[1]
Subsequent NMR, mass spectrometry, chemical degradation, and derivatization experiments enabled the isolation team to identify the key structural features of kedarcidin chromophore, including the enediyne bicyclic core, the ansa-bridging chloropyridyl ring, the mycarose and kedarosamine sugars, and the naphthoamide appendage. However, due to the challenges posed by the complex structure, the initial report had several errors. The bicyclic core proved particularly difficult to deconvolute, as the interpretation of NOE correlations led the researchers to misassign the relative stereochemistry of the core stereotetrad. Moreover, as global absolute chemistry was assigned on the basis of NOE correlations between the stereodefined L-mycarose sugar and the aglycone, the errors of the stereotetrad propagated to the other two stereocenters of the aglycone. Connectivity of the naphthoamide group to the ansa bridge was also misjudged in the initial report.
These errors were later corrected by the independent synthetic efforts of researchers at Tohoku University and Harvard University. In 1997, en route to the originally reported structure, researchers under the direction of Masahiro Hirama discovered that the spectroscopic data of the proposed chloroazatyrosyl (S)-α-amino acid derivative were not consistent with those of the degradation product characterized by Leet et al. Instead, an (R)-β-amino acid derivative was proposed and validated by the Hirama group. This revision led Hirama et al. to invert the other aglycone stereocenters as well, affording a revised structure of kedarcidin chromophore that differed only in the relative stereochemistry of the mycarose-bearing carbon, C10.[2] Finally, in 2007, Myers and co-workers synthesized the structure proposed by Hirama et al.; the corresponding NMR spectroscopic data were distinct from that of the natural product, leading the Myers group to revise the stereochemistry of the mycarose-bearing carbon to 10-(S).[3]

Mechanism of action
Like other enediynes, kedarcidin chromophore comprises a core structure that forms destructive free radicals, as well as appendages that deliver this "warhead" to its DNA target. Thus, the general mechanism by which kedarcidin chromophore damages DNA is known; however, the details of this process—particularly the necessity of nucleophilic activation—have been disputed.
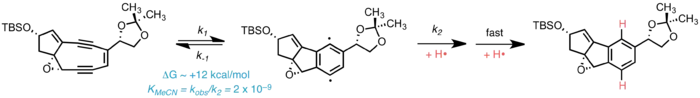
Free-radical DNA damage

The unifying mechanism of bioactivity in all enediyne antibiotics is the Bergman cyclization, wherein the enediyne portion undergoes spontaneous cycloaromatization to generate a para-benzyne biradical activated toward homolytic abstraction of hydrogen from suitable donors, including the deoxyribose sugars of DNA. This generates a carbon-centered free radical on DNA, which undergoes oxidation by molecular oxygen. The resulting peroxide decomposes to form single- or double-stranded breaks in DNA, ultimately leading to cell death.[5]
With considerable sequence selectivity, kedarcidin chromophore binds and cleaves DNA preferentially at TCCTn-mer sites, producing single-strand breaks. Puzzlingly, while the structure of kedarcidin chromophore is most closely related to that of neocarzinostatin chromophore, the former shares sequence-specificity with the structurally distinct calicheamicin enediyne antitumor antibiotic. The naphthoic acid substructure has been implicated in DNA binding, likely through intercalation. To this end, kedarcidin chromophore–induced DNA cleavage is diminished by the addition of divalent cations such as Ca2+ and Mg2+, which chelatively bind the naphthoic acid group of kedarcidin chromophore and thus lessen its affinity for DNA. Competition experiments with netropsin, a known binder of the DNA minor groove, indicate that kedarcidin likely binds the minor groove as well.[6]

Nucleophilic activation
In vivo nucelophilic addition of thiolates to C12 and consequent opening of the core epoxide has been hypothesized to trigger Bergman cyclization in kedarcidin chromophore. Nucleophilic activation is thought to diminish the ring strain incurred by formation of the cycloaromatized product, and thus activate kedarcidin chromophore toward DNA scission.[6] In the isolation and structural characterization studies carried out by Leet et al.,[1] C12-sodium borohydride reduction of kedarcidin chromophore induced rapid cycloaromatization and so facilitated studies of the otherwise unstable natural product. Consequently, C12-nucleophilic activation is proposed extensively in review literature[5] as a possible means for triggering the cycloaromatization event in vivo.

Recent evidence suggests that spontaneous cycloaromatization of kedarcidin chromophore is competitive with nucleophilic bioactivation, if not the predominant mechanism in vivo. While MM2 calculations show that the C1–C12 double bond in the bicyclic core imparts a considerable amount of ring strain (ca. 14 kcal·mol−1) to the [6,5,5] tricycle formed upon Bergman cyclization–reduction, Hirama et al. note that the 5,9-fused enediyne core is susceptible to cycloaromatization–reduction in the absence of both thiol "activating agents" and (non-solvent) hydrogen donors. The kedarcidin chromophore aglycone similarly undergoes reductive cycloaromatization at comparable rates irrespective of the presence of β-mercaptoethanol, a common thiol reductant.[7] In a model system, it was found that the 5,9-bicyclic core of kedarcidin chromophore exists in equilibrium with the corresponding 5,5,6-tricyclic cycloaromatized biradical.[4] The rate of pseudo-first-order decay of this model enediyne is highly dependent on the solvent hydrogen-donor ability, indicating that the hydrogen abstraction step following biradical formation is kinetically significant in the cycloaromatization of the enediyne, as opposed to acyclic systems, where formation of the biradical itself is known to be the rate-limiting step.[8] It is noteworthy that of the solvents examined, tetrahydrofuran—structurally homologous with deoxyribose—led to comparatively fast decomposition of the 5,9-fused enediyne scaffold (t½ = 68 min);[4] Zein et al. independently remark that deoxyribose 4'-hydrogen abstraction is most likely operative in kedarcidin chromophore bioactivity.[6]
Synthesis of epi-kedarcidin chromophore
In 2007, Myers and co-workers at Harvard University reported the synthesis of C10-epi-kedarcidin chromophore, corresponding to the 1997 revised structure advanced by Hirama et al. Critical to the success of this endeavor was retrosynthetic analysis that focused on the convergent coupling of components with roughly equal chemical complexity. Several of the major challenges of C10-epi-kedarcidin chromophore, as well as the strategies used in addressing these difficulties are discussed below.
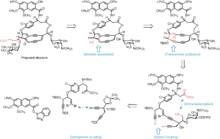
Inherent instability of the enediyne core
The instability toward Bergman cyclization–reduction decomposition pathways poses a major threat to any proposed synthesis of enediynes. Myers and co-workers addressed this liability by late-stage dehydrative installation of the olefin. Without this unsaturation linking the two alkynyl bridges, synthetic intermediates are not disposed toward Bergman-type decomposition, and risk of decomposition is mitigated. In this case, dehydration of a propargylic alcohol was induced by treatment with Martin sulfurane.

Epoxide stereochemistry
In targeting 10-epi-kedarcidin chromophore, Myers et al. sought to install the epoxide functionality syn to the adjacent C10 hydroxyl group. This was accomplished by vanadium-catalyzed epoxidation directed by the C10 hydroxyl group.[9] Had the natural C10-(S)-epimer been desired, it is conceivable that trialkylsilyl protection of the C10 hydroxyl would lead to the desired α-face epoxidation product by steric occlusion of the β face of the olefin; however, without a directing group to accelerate the oxidation of a proximal alkene, this hypothetical reaction would likely suffer from poor regioselectivity, as oxidation of other C–C unsaturations in the molecule would compete with the desired reaction.
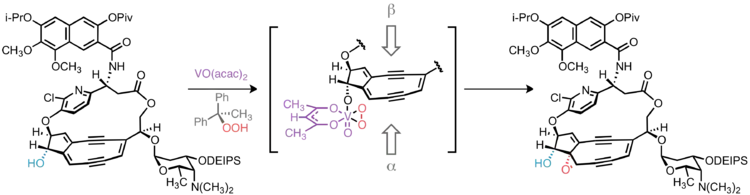
Construction of the bicyclic core
Myers and co-workers have pioneered the application of transannular anionic cyclization reactions in the synthesis of the 5,9-fused bicyclic core of kedarcidin chromophore and neocarzinostatin chromophore. In the first incarnation, hydride delivery to a cyclic tetrayne was guided by aluminum coordination to a proximal alkoxide, thus generating the desired enediyne core in one step via two successive 5-exo-dig–type cyclizations.[10] Later-generation syntheses of the core intercept this cascade cyclization, relying on lithium-halogen exchange on a cyclic vinyl bromide to generate the vinyl anion precursor to the bicyclic product.[3][11]
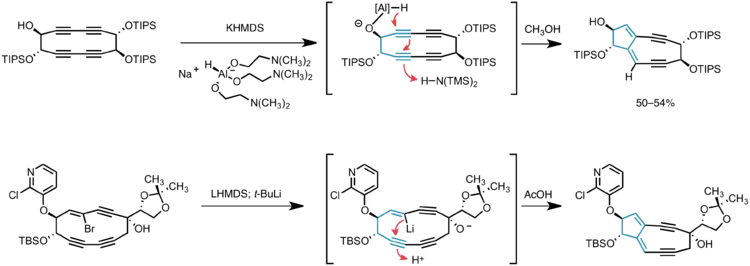
The bicyclic core of C10-epi-kedarcidin chromophore was prepared by the sequential application of three carbon-carbon bond forming reactions, as shown in the retrosynthetic schematic above. First, a Sonogashira coupling was carried out between a bromovinyl electrophile and alkynyl nucleophile; ring closure to give a cyclic triyne was then accomplished by Glaser coupling of two terminal alkynes. The 5,9-fused bicyclic core was established by in situ generation of a vinyllithium species that underwent transannular 5-exo-dig cyclization.
Ansa-bridging macrolactone
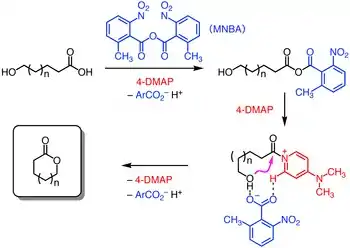
The ansa-bridging macrolactone was constructed following the first Sonogashira coupling, using the Shiina macrolactonization.[12] This protocol was performed on the gram-scale without diminishing its yield employing 2-methyl-6-nitrobenzoic anhydride, 4-dimethylaminopyridine, and triethylamine as a base to promote intramolecular esterification.
Biosynthesis
The means by which bacteria construct enediynes like kedarcidin continues to motivate research. Kedarcidin chromophore, beyond the carbocyclic core it shares with other enediynes, presents additional biosynthetic puzzles: The relative stereochemistry of the groups appended to the carbocyclic core of kedarcidin chomophore differs from that of closely related enediynes; the (R)-2-aza-3-chloro-β-tyrosine substructure has not been identified in any other known natural product; and despite its seeming simplicity, little literature precedence exists for the biosynthesis of the isopropoxy substituent of the naphthonate group.

The biosynthetic gene clusters encoding the biological machinery responsible for producing enediynes have been cloned and characterized for five 9-membered enediynes (C-1027,[13] neocarzinostatin,[14] maduropeptin,[15] sporolides,[16] and kedarcidin[17]), and three 10-membered enediynes (calicheamicin,[18] esperamicin,[19] and dynemicin[20]). Comparative studies of these biosynthetic apparatus have shown that the enediyne core of these molecules is initiated by a common enzyme, enediyne polyketide synthase (PKS). The polyene product of this enzyme is then divergently elaborated into the 9- or 10-membered cores of the enediynes depending on the specific PKS-associated enzymes present. A convergent biosynthetic strategy is then employed by the producing organisms, whereby the varying peripheral appendages of the enediynes are attached to the core structure to furnish the final product.
In 2013, the successful cloning and characterization of the kedarcidin biosynthetic cluster ("ked") was reported by researchers at the Scripps Research Institute and the University of Wisconsin-Madison.[17] The identity of this cloned gene cluster was corroborated by kedA, a gene in the cluster that encodes the previously isolated kedarcidin apoprotein, as well as kedE and kedE10, the co-expression of which in E. coli led to the formation of a signature heptaene product previously implicated in enediyne core biosynthesis.

The 2-aza-β-tyrosine subunit of kedarcidin chromophore is altogether unknown in any other natural product; this lack of precedence frustrates any attempt at a priori identification of the genes responsible for synthesizing this structure. However, six genes are conserved among the biosynthetic clusters of kedarcidin, C-1027, and maduropeptin—while these later two enediynes do not contain a 2-aza-β-tyrosine subunit, they do feature similar (L)-tyrosine-derived components, leading Shen et al. to propose a pathway for the synthesis of the corresponding kedarcidin subunit beginning with 2-aza-L-tyrosine.[17] This α-amino acid is thus believed to be converted to the corresponding β-amino acid by KedY4, an aminomutase encoded in the ked cluster. The resulting product is believed to be loaded onto the peptidyl carrier protein KedY2 and subsequently chlorinated by KedY3, an flavin adenine dinucleotide-dependent halogenase.[17]
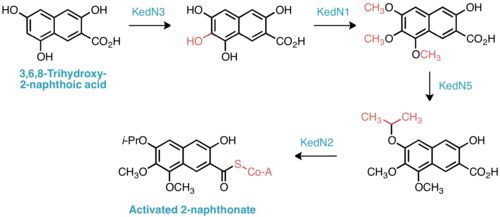
Insight into the biosynthesis of the isopropoxy-2-naphthonate appendage was similarly gained by comparative analysis of the ked cluster to those of neocarzinostatin and maduropeptin, enediynes with naphthonate or benzoate substructures, respectively. Five genes, KedN1–N5, bear high sequence homology with the enzymes responsible for naphthonate synthesis in neocarzinostatin—consequently, the intermediacy of 3,6,8-trihydroxy-2-naphthoic acid is proposed in kedarcidin biosynthesis. This compound is believed to be oxygenated to the 3,6,7,8-tetrahydroxy derivative, then triply O-methylated by KedN1, an O-methyltransferase. To furnish the unique isopropoxy substituent, Shen et al. invoke double C-methylation of the corresponding methoxy group by the radical SAM methyltransferase KedN5.[17]
Conclusion
Owing to its non-specific cytotoxicity, instability under ambient conditions, and relative expense of isolation and manufacture, kedarcidin chromophore has not been investigated rigorously as a therapeutic candidate. However, the recent scientific advances discussed above have served to diminish this last hurdle, as fully synthetic and biosynthetic routes toward scalable kedarcidin production are now within reach. Moreover, with the rising popularity of antibody-drug conjugate therapies, toxicity liabilities may be mitigated through targeted delivery of this potent cytotoxin, potentially enabling efficient therapies that use minimal quantities of this complex material. The recent development of inotuzumab ozogamicin, a calicheamicin-based antibody-drug conjugate for the treatment of non-Hodgkin lymphoma, reinforces the potential of enediynes to find critical use in the treatment of human disease. Thus, the biological potential and complex molecular architecture of kedarcidin may likely inspire further scientific inquiry into this substance, and possibly deliver new ordnance in the war against cancer.
References
- 1 2 3 Leet, J. E.; Schroeder, D. R.; Langley, D. R.; Colson, K. L.; Huang, S.; Klohr, S. E.; Lee, M. S.; Golik, J.; Hofstead, S. J.; Doyle, T. W.; Matson, J. A. J. Am. Chem. Soc. 1993, 115, 8432–8443.
- ↑ Kawata, S.; Ashizawa, S.; Hirama, M. J. Am. Chem. Soc. 1997, 119, 12012–12013.
- 1 2 Ren, F.; Hogan, P. C.; Anderson, A. J.; Myers, A. G. J. Am. Chem. Soc. 2007, 129, 5381–5383.
- 1 2 3 4 Iida, K.-I.; Hirama, M. J. Am. Chem. Soc. 1995, 117, 8875–8876.
- 1 2 (a) Smith, A. L.; Nicolaou, K. C. J. Med. Chem. 1996, 39, 2103. (b) Xi, Z.; Goldberg, I. H. Comp. Nat. Prod. Chem. 1999, 7, 553. (c) Zein, N.; Schroeder, D. R. Adv. DNA Sequence-Specific Agents, 1998, 3, 201.
- 1 2 3 Zein, N.; Colson, K. L.; Leet, J. E.; Schroeder, D. R.; Solomon, W.; Doyle, T. W.; Casazza, A. M. Proc. Natl. Acad. Sci. USA 1993, 90, 2822–2826.
- ↑ Myers, A. G.; Hurd, A. R.; Hogan, P. C. J. Am. Chem. Soc. 2002, 124, 4583–4585.
- ↑ Jones, R. R.; Bergman, R. G. J. Am. Chem. Soc. 1972, 94, 660–661.
- ↑ Rossiter, B. E.; Verhoeven, T. R.; Sharpless, K. B. Tetrahedron Lett. 1979, 20, 4733.
- ↑ Myers, A. G.; Goldberg, S. D. Tetrahedron Lett. 1998, 39, 9633–9636.
- ↑ Myers, A. G.; Goldberg, S. D. Angew. Chem. Int. Ed. 2000, 39, 2732–2735.
- ↑ Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem., 2004, 69, 1822–1830
- ↑ Liu, W.; Christenson, S. D.; Standage, S.; Shen, B. Science 2002, 297, 1170–1173.
- ↑ Liu, W.; Nonaka, K.; Nie, L.; Zhang, J.; Christenson, S. D.; Bae, J.; Van Lanen, S. G.; Zazopoulos, E.; Farnet, C. M.; Yang, C. F.; Shen, B. Chem. Biol. 2005, 12, 293–302.
- ↑ Van Lanen, S. G.; Oh, T.-J.; Liu, W.; Wendt-Pienkowski, E.; Shen, B. J. Am. Chem. Soc. 2007, 129, 13082–13094.
- ↑ McGlinchey, R. P.; Nett, M.; Moore, B. S. J. Am. Chem. Soc. 2008, 130, 2406–2407.
- 1 2 3 4 5 Lohman, J. R.; Huang, S.-X.; Horsman, G. P.; Dilfer, P. E.; Huang, T.; Chen, Y.; Wendt-Pienkowski, E.; Shen, B. Mol. BioSyst. 2013, 9, 478–491.
- ↑ Ahlert, J.; Shepard, E.; Lomovskaya, N.; Zazopoulos, E.; Staffa, A.; Bachmann, B. O.; Huang, K, Fonstein, L.; Czisny, A.; Whitwam, R. E.; Farnet, C. M.; Thorson, T. S. Science 2002, 297, 1173–1176.
- ↑ (a) Zazopoulos, E.; Huang, K.; Staffa, A.; Liu, W.; Bachmann, B. O.; Nonaka, K.; Ahlert, J.; Thorson, J. S.; Shen, B.; Farnet, C. M. Nat. Biotechnol. 2003, 21, 187–190. (b) Liu, W.; Ahlert, J.; Gao, Q.; Wendt-Pienkowski, E.; Shen, B.; Thorson, J. S. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11959–11963.
- ↑ Gao, Q.; Thorson, J. S. FEMS Microbiol. Lett. 2008, 282, 105–114.