| Mucopolysaccharidosis type I | |
|---|---|
| Other names | MPS ! |
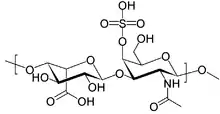 | |
| Structure of dermatan sulfate, one of the molecules that accumulates in the lysosomes of MPS I patients | |
| Causes | Deficiency of the alpha-L iduronidase enzyme |
| Differential diagnosis | Hunter syndrome; other mucopolysaccharidoses |
| Treatment | Enzyme replacement therapy with iduronidase; surgery |
| Prognosis | Death usually occurs before 12 years (Hurler syndrome/severe form); lifespan may be normal (Scheie syndrome/attenuated form) |
| Frequency | 1:100,000 (Hurler syndrome/severe); 1:115,000 (Hurler-Scheie syndrome/intermediate); 1:500,000 (Scheie syndrome/attenuated)[1] |
Mucopolysaccharidosis type I is a spectrum of diseases in the mucopolysaccharidosis family. It results in the buildup of glycosaminoglycans (or GAGs, or mucopolysaccharides) due to a deficiency of alpha-L iduronidase, an enzyme responsible for the degradation of GAGs in lysosomes. Without this enzyme, a buildup of dermatan sulfate and heparan sulfate occurs in the body.
MPS I may present with a wide spectrum of symptoms, depending on how much functional enzyme is produced. In severe forms, symptoms appear during childhood, and early death can occur due to organ damage. In mild cases, the patient may live into adulthood.
Signs and symptoms
MPS I affects multiple organ systems. Children with Hurler syndrome (severe MPS I) may appear normal at birth and develop symptoms over the first years of life. Developmental delay may become apparent by age 1–2 years, with a maximum functional age of 2–4 years. Progressive deterioration follows.
One of the first abnormalities that may be detected is coarsening of the facial features; these symptoms can begin at 3–6 months of age. Skeletal abnormalities occur by about age 6 months, but may not be clinically obvious until 10–14 months. Patients may experience debilitating spine and hip deformities, carpal tunnel syndrome, and joint stiffness. Patients may be normal height in infancy, but stop growing by the age of 2 years. They may not reach a height of greater than 4 feet. Other early symptoms may include inguinal and umbilical hernias. Clouding of the cornea and retinal degeneration may lead to blindness. The liver and spleen may be enlarged due to the deposition of GAGs. Aortic valve disease may occur. Upper and lower respiratory-tract infections can be frequent. Most children develop limited language capabilities. Death usually occurs by age 10.[2][3]
In less severe cases, (Scheie syndrome, or attenuated MPS I), the presentation can vary considerably. Although symptoms generally begin to appear after age 5, the diagnosis is usually made after age 10. Intelligence may be normal, or mild learning disabilities may be present. As with the severe forms, visual problems may lead to blindness. Skeletal deformities and aortic valve disease may occur. These patients may live into adulthood.[1]
Genetics
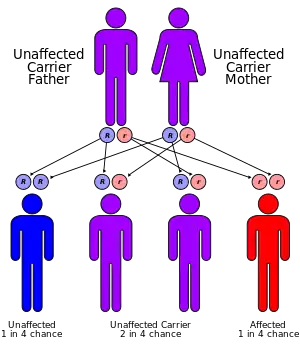
MPS I is inherited in an autosomal recessive manner. This means that patients with MPS I carry two defective copies of the IDUA gene on chromosome 4, with one defective copy being inherited from each parent.
Persons born with one normal copy and one defective copy of the IDUA are called carriers. They produce less α-L-iduronidase than an individual with two normal copies of the gene. The reduced production of the enzyme in carriers, however, remains sufficient for normal function; the person should not show any symptoms of the disease.
Diagnosis
Classification
MPS I is divided into three subtypes based on severity of symptoms. All three types result from an absence of, or insufficient levels of, the enzyme α-L-iduronidase. MPS I H, or Hurler syndrome, is the most severe of the MPS I subtypes. The other two types are MPS I S (Scheie syndrome) and MPS I H-S (Hurler–Scheie syndrome).
Because of the substantial overlap between Hurler syndrome, Hurler-Scheie syndrome, and Scheie syndrome, some sources consider these terms to be outdated. Instead, MPS I may be divided into "severe", "intermediate", and "attenuated" forms.[4][1]
Management
References
- 1 2 3 "Mucopolysaccharidoses Fact Sheet". National Institute of Neurological Disorders and Stroke. 15 Nov 2017. Retrieved 11 May 2018.
- ↑ Banikazemi, Maryam (12 Oct 2014). "Hurler syndrome, Hurler-Scheie Syndrome, and Scheie Syndrome (Mucopolysaccharidosis Type I)". Medscape. Retrieved 10 May 2018.
- ↑ Moore, David; Connock, Martin J.; Wraith, Ed; Lavery, Christine (2008-01-01). "The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK". Orphanet Journal of Rare Diseases. 3: 24. doi:10.1186/1750-1172-3-24. ISSN 1750-1172. PMC 2553763. PMID 18796143.
- ↑ "Mucopolysaccharidosis type I". Genetics Home Reference. Retrieved 10 May 2018.