Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes.[2][3] Gene mapping can also describe the distances between different sites within a gene.
The essence of all genome mapping is to place a collection of molecular markers onto their respective positions on the genome. Molecular markers come in all forms. Genes can be viewed as one special type of genetic markers in the construction of genome maps, and mapped the same way as any other markers. In some areas of study, gene mapping contributes to the creation of new recombinants within an organism.[4]
Gene maps help describe the spatial arrangement of genes on a chromosome. Genes are designated to a specific location on a chromosome known as the locus and can be used as molecular markers to find the distance between other genes on a chromosome. Maps provide researchers with the opportunity to predict the inheritance patterns of specific traits, which can eventually lead to a better understanding of disease-linked traits.[5]
The genetic basis to gene maps is to provide an outline that can potentially help researchers carry out DNA sequencing. A gene map helps point out the relative positions of genes and allows researchers to locate regions of interest in the genome. Genes can then be identified quickly and sequenced quickly.[6]
Two approaches to generating gene maps (gene mapping) include physical mapping and genetic mapping. Physical mapping utilizes molecular biology techniques to inspect chromosomes. These techniques consequently allow researchers to observe chromosomes directly so that a map may be constructed with relative gene positions. Genetic mapping on the other hand uses genetic techniques to indirectly find association between genes. Techniques can include cross-breeding (hybrid) experiments and examining pedigrees. These technique allow for maps to be constructed so that relative positions of genes and other important sequences can be analyzed.[6]
Mapping approaches
There are two distinctive mapping approaches used in the field of genome mapping: genetic maps (also known as linkage maps)[7] and physical maps.[3] While both maps are a collection of genetic markers and gene loci,[8] genetic maps' distances are based on the genetic linkage information, while physical maps use actual physical distances usually measured in number of base pairs. While the physical map could be a more accurate representation of the genome, genetic maps often offer insights into the nature of different regions of the chromosome, for example the genetic distance to physical distance ratio varies greatly at different genomic regions which reflects different recombination rates, and such rate is often indicative of euchromatic (usually gene-rich) vs heterochromatic (usually gene-poor) regions of the genome.
Genetic mapping
Researchers begin a genetic map by collecting samples of blood, saliva, or tissue from family members that carry a prominent disease or trait and family members that do not. The most common sample used in gene mapping, especially in personal genomic tests is saliva. Scientists then isolate DNA from the samples and closely examine it, looking for unique patterns in the DNA of the family members who do carry the disease and the DNA of those who do not carry the disease do not have. These unique molecular patterns in the DNA are referred to as polymorphisms, or markers.[9]
The first steps of building a genetic map are the development of genetic markers and a mapping population. The closer two markers are on the chromosome, the more likely they are to be passed on to the next generation together. Therefore, the "co-segregation" patterns of all markers can be used to reconstruct their order. With this in mind, the genotypes of each genetic marker are recorded for both parents and each individual in the following generations. The quality of the genetic maps is largely dependent upon these factors: the number of genetic markers on the map and the size of the mapping population. The two factors are interlinked, as a larger mapping population could increase the "resolution" of the map and prevent the map from being "saturated".
In gene mapping, any sequence feature that can be faithfully distinguished from the two parents can be used as a genetic marker. Genes, in this regard, are represented by "traits" that can be faithfully distinguished between two parents. Their linkage with other genetic markers is calculated in the same way as if they are common markers and the actual gene loci are then bracketed in a region between the two nearest neighboring markers. The entire process is then repeated by looking at more markers that target that region to map the gene neighborhood to a higher resolution until a specific causative locus can be identified. This process is often referred to as "positional cloning", and it is used extensively in the study of plant species. One plant species, in particular in which positional cloning is utilized is in maize.[10] The great advantage of genetic mapping is that it can identify the relative position of genes based solely on their phenotypic effect.
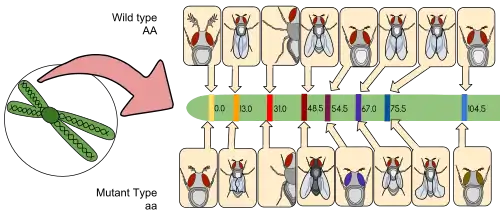
Genetic mapping is a way to identify exactly which chromosome has which gene and exactly pinpointing where that gene lies on that particular chromosome. Mapping also acts as a method in determining which gene is most likely to recombine based on the distance between two genes. The distance between two genes is measured in units known as centimorgan or map units, these terms are interchangeable. A centimorgan is a distance between genes for which one product of meiosis in one hundred is recombinant.[11][4] The farther two genes are from each other, the more likely they are going to recombine. If it were closer, the opposite would occur.[12]
Linkage analysis
The basis to linkage analysis is understanding chromosomal location and identifying disease genes. Certain genes that are genetically linked or associated with each other reside close to each other on the same chromosome. During meiosis, these genes are capable of being inherited together and can be used as a genetic marker to help identify the phenotype of diseases. Because linkage analysis can identify inheritance patterns, these studies are usually family based.[13]
Gene association analysis
Gene association analysis is population based; it is not focused on inheritance patterns, but rather is based on the entire history of a population. Gene association analysis looks at a particular population and tries to identify whether the frequency of an allele in affected individuals is different from that of a control set of unaffected individuals of the same population. This method is particularly useful to identify complex diseases that do not have a Mendelian inheritance pattern.[14]
Physical mapping
Since actual base-pair distances are generally hard or not possible to directly measure, physical maps are actually constructed by first shattering the genome into hierarchically smaller pieces. By characterizing each single piece and assembling back together, the overlapping path or "tiling path" of these small fragments would allow researchers to infer physical distances between genomic features.
Restriction mapping is a method in which structural information regarding a segment of DNA is obtained using restriction enzymes. Restriction enzymes are enzymes that help cut segments of DNA at specific recognition sequences. The basis to restriction mapping involves digesting (or cutting) DNA with restriction enzymes. The digested DNA fragments are then run on an agarose gel using electrophoresis, which provides one with information regarding the size of these digested fragments. The sizes of these fragments help indicate the distance between restriction enzyme sites on the DNA analyzed, and provides researchers with information regarding the structure of DNA analyzed.[14] The resulting pattern of DNA migration – its genetic fingerprint is used to identify what stretch of DNA is in the clone. By analyzing the fingerprints, contigs are assembled by automated (FPC) or manual means (pathfinders) into overlapping DNA stretches. Now a good choice of clones can be made to efficiently sequence the clones to determine the DNA sequence of the organism under study.
In physical mapping, there are no direct ways of marking up a specific gene since the mapping does not include any information that concerns traits and functions. Genetic markers can be linked to a physical map by processes like in situ hybridization. By this approach, physical map contigs can be "anchored" onto a genetic map. The clones used in the physical map contigs can then be sequenced on a local scale to help new genetic marker design and identification of the causative loci.
Macrorestriction is a type of physical mapping wherein the high molecular weight DNA is digested with a restriction enzyme having a low number of restriction sites.
There are alternative ways to determine how DNA in a group of clones overlaps without completely sequencing the clones. Once the map is determined, the clones can be used as a resource to efficiently contain large stretches of the genome. This type of mapping is more accurate than genetic maps.
Restriction mapping
Restriction mapping is a method in which structural information regarding a segment of DNA is obtained using restriction enzymes. Restriction enzymes are enzymes that help cut segments of DNA at specific recognition sequences. The basis to restriction mapping involves digesting (or cutting) DNA with restriction enzymes. The digested DNA fragments are then run on an agarose gel using electrophoresis, which provides one with information regarding the size of these digested fragments. The sizes of these fragments help indicate the distance between restriction enzyme sites on the DNA analyzed, and provides researchers with information regarding the structure of DNA analyzed.[14]
Fluorescent in situ hybridization
Fluorescence in situ hybridization (FISH) is a method used to detect the presence (or absence) of a DNA sequence within a cell.[15] DNA probes that are specific for chromosomal regions or genes of interest are labeled with fluorochromes. By attaching fluorochromes to probes, researchers are able to visualize multiple DNA sequences simultaneously. When a probe comes into contact with DNA on a specific chromosome, hybridization will occur. Consequently, information regarding the location of that sequence of DNA will be attained. FISH analyzes single stranded DNA (ssDNA). Once the DNA is in its single stranded state, the DNA can bind to its specific probe.[6]
Sequence-tagged site (STS) mapping
A sequence-tagged site (STS) is a short sequence of DNA (about 100 - 500 base pairs in length) that is seen to appear multiple times within an individual's genome. These sites are easily recognizable, usually appearing at least once in the DNA being analyzed. These sites usually contain genetic polymorphisms making them sources of viable genetic markers (as they differ from other sequences). Sequenced tagged sites can be mapped within our genome and require a group of overlapping DNA fragments. PCR is generally used to produce the collection of DNA fragments. After overlapping fragments are created, the map distance between STSs can be analyzed. In order to calculate the map distance between STSs, researchers determine the frequency at which breaks between the two markers occur (see shotgun sequencing)[14]
Mapping mutational sites
In the early 1950s the prevailing view was that the genes in a chromosome are discrete entities, indivisible by genetic recombination and arranged like beads on a string. During 1955 to 1959, Benzer performed genetic recombination experiments using rII mutants of bacteriophage T4. He found that, on the basis of recombination tests, the sites of mutation could be mapped in a linear order.[16][17] This result provided evidence for the key idea that the gene has a linear structure equivalent to a length of DNA with many sites that can independently mutate.
In 1961, Francis Crick, Leslie Barnett, Sydney Brenner and Richard Watts-Tobin performed genetic experiments that demonstrated the basic nature of the genetic code for proteins.[18] These experiments, involving mapping of mutational sites within the rIIB gene of bacteriophage T4, demonstrated that three sequential nucleobases of the gene's DNA specify each successive amino acid of its encoded protein. Thus the genetic code was shown to be a triplet code, where each triplet (called a codon) specifies a particular amino acid. They also obtained evidence that the codons do not overlap with each other in the DNA sequence encoding a protein, and that such a sequence is read from a fixed starting point.
Edgar et al.[19] performed mapping experiments with r mutants of bacteriophage T4 showing that recombination frequencies between rII mutants are not strictly additive. The recombination frequency from a cross of two rII mutants (a x d) is usually less than the sum of recombination frequencies for adjacent internal sub-intervals (a x b) + (b x c) + (c x d). Although not strictly additive, a systematic relationship was demonstrated[20] that likely reflects the underlying molecular mechanism of genetic recombination.
Genome sequencing
Genome sequencing is sometimes mistakenly referred to as "genome mapping" by non-biologists. The process of shotgun sequencing[21] resembles the process of physical mapping: it shatters the genome into small fragments, characterizes each fragment, then puts them back together (more recent sequencing technologies are drastically different). While the scope, purpose and process are totally different, a genome assembly can be viewed as the "ultimate" form of physical map, in that it provides in a much better way all the information that a traditional physical map can offer.
Use
Identification of genes is usually the first step in understanding a genome of a species; mapping of the gene is usually the first step of identification of the gene. Gene mapping is usually the starting point of many important downstream studies.
Disease association
The process to identify a genetic element that is responsible for a disease is also referred to as "mapping". If the locus in which the search is performed is already considerably constrained, the search is called the fine mapping of a gene. This information is derived from the investigation of disease manifestations in large families (genetic linkage) or from populations-based genetic association studies.
Using the methods mentioned above, researchers are capable of mapping disease genes. Generating a gene map is the critical first step towards identifying disease genes. Gene maps allow for variant alleles to be identified and allow for researchers to make predictions about the genes they think are causing the mutant phenotype. An example of a disorder that was identified by Linkage analysis is Cystic Fibrosis. For example, with Cystic Fibrosis (CF), DNA samples from fifty families affected by CF were analyzed using linkage analysis. Hundreds of markers pertaining to CF were analyzed throughout the genome until CF was identified on the long arm of chromosome 7. Researchers then had completed linkage analysis on additional DNA markers within chromosome 7 to identify an even more precise location of the CF gene. They found that the CF gene resides around 7q31-q32 (see chromosomal nomenclature).[14]
See also
References
- ↑ Mader S (2007). Biology (Ninth ed.). New York: McGraw-Hill. p. 209. ISBN 978-0-07-325839-3.
- ↑ "Gene mapping - Glossary Entry". Genetics Home Reference]. Bethesda, MD: Lister Hill National Center for Biomedical Communications, an Intramural Research Division of the U.S. National Library of Medicine. 2013-09-03. Retrieved 2013-09-06.
- 1 2 "Mapping". Genome.gov. Retrieved 3 May 2023.
- 1 2 Ladejobi O, Elderfield J, Gardner KA, Gaynor RC, Hickey J, Hibberd JM, et al. (December 2016). "Maximizing the potential of multi-parental crop populations". Applied & Translational Genomics. 11: 9–17. doi:10.1016/j.atg.2016.10.002. PMC 5167364. PMID 28018845.
- ↑ Nussbaum, Robert L.; McInnes, Roderick R.; Wilard, Huntington F. (2016). Thompson & Thompson Genetics in Medicine (Eighth ed.). Philadelphia, PA: Elsevier. pp. 178–187. ISBN 978-1-4377-0696-3. Archived from the original on 4 March 2016. Retrieved 13 October 2015.
- 1 2 3 Brown, Terence, A. (2002). Genomes. Manchester, UK: Garland Science. ISBN 0-471-25046-5.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ↑ "Genetic Map". Genome.gov. Retrieved 2 May 2023.
- ↑ Aguilera-Galvez C, Champouret N, Rietman H, Lin X, Wouters D, Chu Z, et al. (March 2018). "Two different R gene loci co-evolved with Avr2 of Phytophthora infestans and confer distinct resistance specificities in potato". Studies in Mycology. 89: 105–115. doi:10.1016/j.simyco.2018.01.002. PMC 6002340. PMID 29910517.
- ↑ "Genetic Mapping Fact Sheet".
- ↑ Gallavotti A, Whipple CJ (January 2015). "Positional cloning in maize (Zea mays subsp. mays, Poaceae)". Applications in Plant Sciences. 3 (1): 1400092. doi:10.3732/apps.1400092. PMC 4298233. PMID 25606355.
- ↑ Saygin D, Tabib T, Bittar HE, Valenzi E, Sembrat J, Chan SY, et al. (2017-04-01). "Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension". Pulmonary Circulation. Advances in Crop Science: Innovation and Sustainability. 10 (1): 175–184. doi:10.1016/j.cj.2016.06.003. PMC 7052475. PMID 32166015.
- ↑ Goldberg M, Fischer J, Hood L, Hartwell L (2020). Genetics: From Genes to Genomes. New York, NY: McGraw Hill. pp. 125–128. ISBN 978-1-260-24087-0.
- ↑ Pulst, Stefan M. (June 1999). "Genetic Linkage Analysis". JAMA Neurology. 56 (6): 667–672. doi:10.1001/archneur.56.6.667. PMID 10369304. Retrieved 13 October 2015.
- 1 2 3 4 5 Hartwell, Leland H.; Hood, Leroy; Goldberg, Michael L.; Reynolds, Anne E.; Silver, Lee M.; Karagiannis, Jim; Papaconstantinou, Maria (2014). Genetics: From Genes to Genomes (Canadian ed.). Canada: McGraw-Hill Ryerson. pp. 456–459, 635–636. ISBN 978-0-07-094669-9. Retrieved 13 October 2015.
- ↑ "Fluorescence In Situ Hybridization Fact Sheet". Genome.gov. Retrieved 3 May 2023.
- ↑ Benzer S (June 1955). "Fine structure of a genetic region in bacteriophage". Proceedings of the National Academy of Sciences of the United States of America. 41 (6): 344–54. doi:10.1073/pnas.41.6.344. PMC 528093. PMID 16589677.
- ↑ Benzer S (November 1959). "On the topology of the genetic fine structure". Proceedings of the National Academy of Sciences of the United States of America. 45 (11): 1607–20. doi:10.1073/pnas.45.11.1607. PMC 222769. PMID 16590553.
- ↑ Crick FH, Barnett L, Brenner S, Watts-Tobin RJ (December 1961). "General nature of the genetic code for proteins". Nature. 192: 1227–32. doi:10.1038/1921227a0. PMID 13882203.
- ↑ Edgar RS, Feynman RP, Klein S, Lielausis I, Steinberg CM (February 1962). "Mapping experiments with r mutants of bacteriophage T4D". Genetics. 47 (2): 179–186. doi:10.1093/genetics/47.2.179. PMC 1210321. PMID 13889186.
- ↑ Fisher KM, Bernstein H (December 1965). "The additivity of intervals in the RIIA cistron of phage T4D". Genetics. 52 (6): 1127–36. doi:10.1093/genetics/52.6.1127. PMC 1210971. PMID 5882191.
- ↑ Saygin D, Tabib T, Bittar HE, Valenzi E, Sembrat J, Chan SY, et al. (2018). "Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension". Pulmonary Circulation. 10 (1): 890–898. doi:10.1080/1828051X.2018.1462110. PMC 7052475. PMID 32166015.
Further reading
- Brown TA (2007). Genomes 3. New York, NY: Garland Science Publishing. ISBN 9780815341383. OCLC 444522997.
External links
- "Genetic Mapping Fact Sheet". Bethesda, MD: National Human Genome Research Institute, National Institutes of Health. Retrieved 2013-09-06.
- "Canada's Michael Smith Genome Sciences Centre". Vancouver, British Columbia. Retrieved 2013-09-06.