Mutational signatures are characteristic combinations of mutation types arising from specific mutagenesis processes such as DNA replication infidelity, exogenous and endogenous genotoxin exposures, defective DNA repair pathways, and DNA enzymatic editing.[1]
The term is used for two distinct concepts, often conflated: mutagen signatures and tumor signatures. Its original use, mutagen signature, referred to a pattern of mutations made in the laboratory by a known mutagen and not made by other mutagens – unique to the mutagen as a human signature is unique to the signer. Uniqueness allows the mutagen to be deduced from a cell's mutations [2] Later, the phrase referred to a pattern of mutations characteristic of a tumor type, although usually not unique to the tumor type nor to a mutagen.[3][4] If a tumor mutational signature matches a unique mutagen mutational signature, it is valid to deduce the carcinogen exposure or mutagenesis process that occurred in the patient's distant past.[2] Increasingly refined tumor signatures are becoming assignable to mutagen signatures.[5]
Deciphering mutational signatures in cancer provides insight into the biological mechanisms involved in carcinogenesis and normal somatic mutagenesis.[6] Mutational signatures have shown their applicability in cancer treatment and cancer prevention. Advances in the fields of oncogenomics have enabled the development and use of molecularly targeted therapy, but such therapies historically focused on inhibition of oncogenic drivers (e.g. EGFR gain-of-function mutation and EGFR inhibitor treatment in colorectal cancer[7]). More recently, mutational signatures profiling has proven successful in guiding oncological management and use of targeted therapies (e.g. immunotherapy in mismatch repair deficient of diverse cancer types,[8] platinum and PARP inhibitor to exploit synthetic lethality in homologous recombination deficient breast cancer).[9]
General concepts
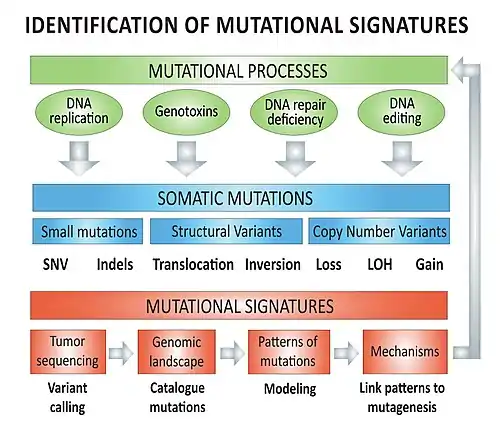
Mechanisms – overview
The biological mutagenesis mechanisms underlying mutational signatures (e.g. COSMIC Signatures 1 to 30) include, but are not limited to:[lower-alpha 1][4]
- DNA replication infidelity
- DNA proofreading is the process by which DNA polymerase excises an incorrectly incorporated nucleotide via exonuclease enzymatic reaction. Inability of DNA polymerase to correct these replication errors leads to progressive accumulation of mutations through successive cell mitosis.
- Genotoxins
- Endogenous cellular (e.g. spontaneous 5-methylcytosine deamination leads to C>T transition (genetics)) mutations (see DNA damage (naturally occurring))
- Exogenous/carcinogens
- Ultraviolet radiation: UVB radiation causes direct DNA damage and is a known risk factor for skin cancer (e.g. melanoma)
- Alkylating antineoplastic agents: This group of chemotherapy agents adds alkyl group to DNA, which causes crosslinking of DNA and interferes with DNA replication and DNA repair. Cancer cells are most impacted because of their high mitosis rate.
- Tobacco: Tobacco contains several carcinogens which are harmful to DNA, including polycyclic aromatic hydrocarbons, acrolein, nitrosamines, cyanide and others (see health effects of tobacco)
- DNA repair deficiency
- Homologous recombination deficiency (HRD): DNA double-strand break requires homologous recombination mechanism for accurate repair of breakpoints.
- DNA mismatch repair (MMR) deficiency: The mismatch repair machinery recognizes and repairs erroneous base pair insertion, deletion or mis-incorporation.
- Enzymatic DNA editing
- Cytidine deaminase enzymes: This family of enzymes are part of the innate immune system and are involved in the control of retroviruses and transposons elements (including endogenous retroviruses). These enzymes (cytidine deaminase/CDA, activation-induced cytidine deaminase and APOBEC protein family) actively cause cytidine deamination and therefore introduce C>T transition (genetics) mutations.
- DNA replication infidelity
Genomic data
Cancer mutational signatures analyses require genomic data from cancer genome sequencing with paired-normal DNA sequencing in order to create the tumor mutation catalog (mutation types and counts) of a specific tumor. Different types of mutations (e.g. single nucleotide variants, indels, structural variants) can be used individually or in combination to model mutational signatures in cancer.
Types of mutations: base substitutions
There are six classes of base substitution: C>A, C>G, C>T, T>A, T>C, T>G. The G>T substitution is considered equivalent to the C>A substitution because it is not possible to differentiate on which DNA strand (forward or reverse) the substitution initially occurred. Both the C>A and G>T substitutions are therefore counted as part of the "C>A" class. For the same reason the G>C, G>A, A>T, A>G and A>C mutations are counted as part of the "C>G", "C>T", "T>A", "T>C" and "T>G" classes respectively.
Taking the information from the 5' and 3' adjacent bases (also called flanking base pairs or trinucleotide context) lead to 96 possible mutation types (e.g. A[C>A]A, A[C>A]T, etc.). The mutation catalog of a tumor is created by categorizing each single nucleotide variant (SNV) (synonyms: base-pair substitution or substitution point mutation) in one of the 96 mutation types and counting the total number of substitutions for each of these 96 mutation types (see figure).
Tumor mutation catalog

Once the mutation catalog (e.g. counts for each of the 96 mutation types) of a tumor is obtained, there are two approaches to decipher the contributions of different mutational signatures to tumor genomic landscape:
- The mutation catalog of the tumor is compared to a reference mutation catalogue, or mutational signatures reference dataset, such as the 21 Signatures of Mutational Processes in Human Cancer [4] from the Catalogue of Somatic Mutation In Cancer (COSMIC) database.[1]
- De novo mutational signatures modelling can be accomplished using statistical methods such as non-negative matrix factorization to identify potential novel mutational processes.[10]
Identifying the contributions of diverse mutational signatures to carcinogenesis provides insight into tumor biology and can offer opportunities for targeted therapy.
Types of mutations: indels
Signature 3, seen in homologous recombination (HR) deficient tumour, is associated with increased burden of large indels (up to 50 nucleotides) with overlapping microhomology at the breakpoints.[4] In such tumors, DNA double-strand breaks are repaired by the imprecise repair mechanisms of non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ) instead of high fidelity HR repair.
Signature 6, seen in tumors with microsatellite instability, also features enrichment of 1bp indels in nucleotide repeat regions.
Types of mutations: structural variants
Homologous recombination deficiency leads to Signature 3 substitution pattern, but also to increase burden of structural variants. In the absence of homologous recombination, non-homologous end joining leads to large structural variants such as chromosomal translocations, chromosomal inversions and copy number variants.
Mutational signatures
A brief description of selected mutational processes and their associated mutational signatures in cancer will be included in the sections below. Some signatures are ubiquitous across diverse cancer types (e.g. Signature 1) while some others tend to associate with specific cancers (e.g. Signature 9 and lymphoid malignancies).[4]
Some mutational signatures feature strong transcriptional-bias with substitutions preferentially affecting one of the DNA strands, either the transcribed or untranscribed strand (Signatures 5, 7, 8, 10, 12, 16).[4]
Age-related mutagenesis
Signature 1 features a predominance of C>T transition (genetics) in the Np[C>T]G trinucleotide contexts and correlates with the age of patient at time of cancer diagnosis. The underlying proposed biological mechanism is the spontaneous deamination of 5-methylcytosine.[4]
Signature 5 has a predominance of T>C substitutions in the ApTpN trinucleotide context with transcriptional strand bias.[6]
Homologous recombination deficiency
Signature 3 displays high mutation counts of multiple mutation classes and is associated with germline and somatic (biology) BRCA1 and BRCA2 mutations in several cancer types (e.g. breast, pancreatic, ovarian, prostate). This signature results from DNA double-strand break repair deficiency (or homologous recombination deficiency). Signature 3 is associated with high burden of indels with microhomology at the breakpoints.[6]
APOBEC enzymes
APOBEC3 family of cytidine deaminase enzymes respond to viral infections by editing viral genome, but the enzymatic activity of APOBEC3A and APOBEC3B has also been found to cause unwanted host genome editing and may even participate to oncogenesis in human papillomavirus-related cancers.[11]
Signature 2 and Signature 13 are enriched for C>T and C>G substitutions and are thought to arise from cytidine deaminase activity of the AID/APOBEC enzymes family.[6]
A germline deletion polymorphism involving APOBEC3A and APOBEC3B is associated with high burden of Signature 2 and Signature 13 mutations.[12] This polymorphism is considered to be of moderate penetrance (two-fold above background risk) for breast cancer risk.[13] The exact roles and mechanisms underlying APOBEC-mediated genome editing are not yet fully delineated, but activation-induced cytidine deaminase(AID)/APOBEC complex is thought to be involved in host immune response to viral infections and lipid metabolism.[14]
Both Signature 2 and Signature 13 feature cytosine to uracil substitutions due to cytidine deaminases. Signature 2 has a higher proportion of C[T>C]N substitutions and Signature 13 a higher proportion of T[C>G]N substitutions. APOBEC3A and APOBEC3B-mediated mutagenesis preferentially involve the lagging DNA strand during replication.[15]
Mismatch repair deficiency
Four COSMIC mutational signatures have been associated with DNA mismatch repair deficiency and found in tumors with microsatellite instability: Signature 6, 15, 20 and 26.[6] Loss of function MLH1, MSH2, MSH6 or PMS2 genes cause defective DNA mismatch repair.
DNA proofreading
Signature 10 has a transcriptional bias and is enriched for C>A substitutions in the TpCpT context as well as T>G substitutions in the TpTpTp context.[6] Signature 10 is associated with altered function of DNA polymerase epsilon, which result in deficient DNA proofreading activity. Both germline and somatic POLE (gene) exonuclease domain mutations are associated with Signature 10.[16]
Base excision repair

Somatic enrichment for transversion mutations (G:C>T:A) has been associated with base excision repair (BER) deficiency and linked to defective MUTYH, a DNA glycosylase, in colorectal cancer.[17] Direct DNA oxidation damage leads to the creation of 8-Oxoguanine, which if remains un-repaired, will lead to incorporation of adenine instead of cytosine during DNA replication. MUTYH encodes the mutY adenine glycosylase enzyme which excise the mismatched adenine from 8-Oxoguanine:adenine base pairing, therefore enabling DNA repair mechanisms involving OGG1 (Oxoguanine glycosylase) and NUDT1 (Nudix hydrolase 1, also known as MTH1, MutT homolog 1) to remove the damaged 8-Oxoguanine.[18]
Exposures to exogenous genotoxins
Selected exogenous genotoxins/carcinogens and their mutagen-induced DNA damage and repair mechanisms have been linked to specific molecular signatures.
Ultraviolet radiation (UV)
- Signature 7 has a predominance of C>T substitutions at sites of adjacent pyrimidines (adjacent C or T), with a particularly diagnostic subset being the CC>TT dinucleotide mutation. This pattern arises because the major UV-induced DNA photoproducts join two adjacent pyrimidines; the photoproduct is typically the cyclobutane pyrimidine dimer (CPD).[19] Specificity for C>T appears to be due to the million-fold acceleration of C deamination when it is part of a CPD, with the resulting uracil acting as T.[20][21] CPDs are repaired via transcription-coupled nucleotide excision repair, causing a strong bias for C>T substitutions enriched on the untranscribed DNA strand.[6] The regions of a tumor suppressor protein that are mutationally inactivated in sunlight-related skin cancers are the same as in cancers of organs not exposed to sunlight, but the nucleotide mutated is often shifted a few bases to a site where a CPD could form.[22] Ultraviolet radiation exposure is therefore the proposed underlying mutagenic mechanism of this signature. UV also illustrates a subtlety in interpreting a tumor signature as a mutagen signature: only three-quarters of mutations induced by UV in the laboratory are UV signature mutations because UV also triggers cellular oxidative processes.[2] Therefore even if all mutations in a tumor were caused by UV from sunlight, one quarter of the mutations are expected to not be UV signature mutations. A second carcinogen needn't be invoked to explain those mutations, but a second mutational process is required. Identification of a UV signature in a tumor of unknown primary site is clinically important as it suggests a diagnosis of metastatic skin cancer and has important treatment implications.[23]
Alkylating agents
- Signature 11 was identified in tumors previously exposed to Temozolamide, an alkylating agent.[6] This signature is enriched for C>T substitutions on guanine bases due to transcription-coupled nucleotide excision repair. A strong transcriptional strand-bias is present in this signature.
Tobacco
- Both Signature 4 (tobacco smoking, lung cancer) and Signature 29 (tobacco chewing, gingivo-buccal oral squamous cell carcinoma) display transcriptional strand-bias and enrichment for C>A substitutions, but their respective composition and patterns (proportion of each mutation types) differ slightly.[6]
- The proposed underlying mechanism of Signature 4 is the removal of DNA adducts (tobacco benzo(a)pyrene covalently bounded to guanine) by the transcription-coupled nucleotide excision repair (NER) machinery.[24]
Immunoglobulin gene hypermutation
Signature 9 has been identified in chronic lymphocytic leukemia and malignant B-cell lymphoma and feature enrichment for T>G transversion events. It is thought to result from error-prone polymerase η (POLH gene)-associated mutagenesis.[4]
Recently, polymerase η error-prone synthesis signature has been linked to non-hematological cancers (e.g. skin cancer) and was hypothesized to contribute to YCG motif mutagenesis and could partly explain the increase TC dinucleotides substitutions.[25]
History
During the 1990s, Curtis Harris at the US National Cancer Institute and Bert Vogelstein at the Johns Hopkins Oncology Center in Baltimore reviewed data showing that different types of cancer had their own unique suite of mutations in p53, which were likely to have been caused by different agents,[3][26] such as the chemicals in tobacco smoke or ultraviolet light from the sun.[19][27] With the advent of next-generation sequencing, Michael Stratton saw the potential for the technology to revolutionize our understanding of the genetic changes inside individual tumors, setting the Wellcome Sanger Institute's huge banks of DNA-sequencing machines in motion to read every single letter of DNA in a tumor.[28] By 2009, Stratton and his team had produced the first whole cancer genome sequences. These were detailed maps showing all the genetic changes and mutations that had occurred within two individual cancers—a melanoma from the skin and a lung tumor.[29][30] The melanoma and lung cancer genomes were powerful proof that the fingerprints of specific culprits could be seen in cancers with one major cause. These tumors still contained many mutations that could not be explained by ultraviolet light or tobacco smoking. The detective work became a lot more complicated for cancers with complex, multiple or even completely unknown origins. By way of analogy, imagine a forensic scientist dusting for fingerprints at a murder scene. The forensic scientist might strike it lucky and find a set of perfect prints on a windowpane or door handle that match a known killer. However, they are much more likely to uncover a mish-mash of fingerprints belonging to a whole range of folk—from the victim and potential suspects to innocent parties and police investigators—all laid on top of each other on all sorts of surfaces.[28] This is very similar to cancer genomes where multiple mutational patterns are commonly overlaid one over another making the data incomprehensible. Fortunately, a PhD student of Stratton's, Ludmil Alexandrov came up with a way of mathematically solving the problem. Alexandrov demonstrated that mutational patterns from individual mutagens found in a tumor can be distinguished from one another using a mathematical approach called blind source separation. The newly disentangled patterns of mutations were termed mutational signatures.[28] In 2013, Alexandrov and Stratton published the first computational framework for deciphering mutational signatures from cancer genomics data.[31] Subsequently, they applied this framework to more than seven thousand cancer genomes creating the first comprehensive map of mutational signatures in human cancer.[32] Currently, more than one hundred mutational signatures have been identified across the repertoire of human cancer.[33] In April 2022 58 new mutational signatures were described.[34][35][36]
See also
Note list
- ↑ As DNA replication, maintenance and repair is not a linear process, some signatures are caused by overlapping mutagenesis mechanisms.
References
- 1 2 Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. (January 2017). "COSMIC: somatic cancer genetics at high-resolution". Nucleic Acids Research. 45 (D1): D777–D783. doi:10.1093/nar/gkw1121. PMC 5210583. PMID 27899578.
- 1 2 3 Brash DE (2015). "UV signature mutations". Photochemistry and Photobiology. 91 (1): 15–26. doi:10.1111/php.12377. PMC 4294947. PMID 25354245.
- 1 2 Hollstein M, Sidransky D, Vogelstein B, Harris CC (July 1991). "p53 mutations in human cancers". Science. 253 (5015): 49–53. Bibcode:1991Sci...253...49H. doi:10.1126/science.1905840. PMID 1905840. S2CID 38527914.
- 1 2 3 4 5 6 7 8 9 10 Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. (August 2013). "Signatures of mutational processes in human cancer" (PDF). Nature. 500 (7463): 415–21. Bibcode:2013Natur.500..415.. doi:10.1038/nature12477. PMC 3776390. PMID 23945592.
- ↑ Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, Gomez C, Degasperi A, Harris R, Jackson SP, Arlt VM, Phillips DH, Nik-Zainal S (2019). "A compendium of mutational signatures of environmental agents". Cell. 177 (4): 821-36 e16. doi:10.1016/j.cell.2019.03.001. PMC 6506336. PMID 30982602.
- 1 2 3 4 5 6 7 8 9 Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR (December 2015). "Clock-like mutational processes in human somatic cells". Nature Genetics. 47 (12): 1402–7. doi:10.1038/ng.3441. PMC 4783858. PMID 26551669.
- ↑ Seow H, Yip WK, Fifis T (March 2016). "Advances in Targeted and Immunobased Therapies for Colorectal Cancer in the Genomic Era". OncoTargets and Therapy. 9 (9): 1899–920. doi:10.2147/OTT.S95101. PMC 4821380. PMID 27099521.
- ↑ Chuk MK, Chang JT, Theoret MR, Sampene E, He K, Weis SL, Helms WS, Jin R, Li H, Yu J, Zhao H, Zhao L, Paciga M, Schmiel D, Rawat R, Keegan P, Pazdur R (October 2017). "FDA Approval Summary: Accelerated Approval of Pembrolizumab for Second-Line Treatment of Metastatic Melanoma". Clinical Cancer Research. 23 (19): 5666–5670. doi:10.1158/1078-0432.CCR-16-0663. PMID 28235882.
- ↑ O'Neil, Nigel J.; Bailey, Melanie L.; Hieter, Philip (26 June 2017). "Synthetic lethality and cancer". Nature Reviews Genetics. 18 (10): 613–623. doi:10.1038/nrg.2017.47. PMID 28649135. S2CID 3422717.
- 1 2 Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, et al. (December 2017). "Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer". Clinical Cancer Research. 23 (24): 7521–7530. doi:10.1158/1078-0432.CCR-17-1941. PMID 29246904.
- ↑ Warren C, Westrich J, Doorslaer K, Pyeon D (August 2017). "Roles of APOBEC3A and APOBEC3B in Human Papillomavirus Infection and Disease Progression". Viruses. 9 (8): 233. doi:10.3390/v9080233. PMC 5580490. PMID 28825669.
- ↑ Middlebrooks CD, Banday AR, Matsuda K, Udquim KI, Onabajo OO, Paquin A, et al. (November 2016). "Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors". Nature Genetics. 48 (11): 1330–1338. doi:10.1038/ng.3670. PMC 6583788. PMID 27643540.
- ↑ Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, et al. (May 2014). "Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer". Nature Genetics. 46 (5): 487–91. doi:10.1038/ng.2955. PMC 4137149. PMID 24728294.
- ↑ Yang B, Li X, Lei L, Chen J (September 2017). "APOBEC: From mutator to editor". Journal of Genetics and Genomics = Yi Chuan Xue Bao. 44 (9): 423–437. doi:10.1016/j.jgg.2017.04.009. PMID 28964683.
- ↑ Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA (February 2016). "APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication". Cell Reports. 14 (6): 1273–1282. doi:10.1016/j.celrep.2016.01.021. PMC 4758883. PMID 26832400.
- ↑ Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, Church DN (February 2016). "A panoply of errors: polymerase proofreading domain mutations in cancer". Nature Reviews. Cancer. 16 (2): 71–81. doi:10.1038/nrc.2015.12. PMID 26822575. S2CID 9359891.
- 1 2 Viel, A, Bruselles, A, Meccia, E, et al. (April 2017). "A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer". eBioMedicine. 20: 39–49. doi:10.1016/j.ebiom.2017.04.022. PMC 5478212. PMID 28551381.
- ↑ David, SS; O'Shea, VL; Kundu, S (2007). "Base-excision repair of oxidative DNA damage". Nature. 447 (7147): 941–950. Bibcode:2007Natur.447..941D. doi:10.1038/nature05978. PMC 2896554. PMID 17581577.
- 1 2 Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Pontén J (1991). "A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma". Proceedings of the National Academy of Sciences USA. 88 (22): 10124–8. Bibcode:1991PNAS...8810124B. doi:10.1073/pnas.88.22.10124. PMC 52880. PMID 1946433.
- ↑ Cannistraro VJ, Pondugula S, Song Q, Taylor JS (2015). "Rapid deamination of cyclobutane pyrimidine dimer photoproducts at TCG sites in a translationally and rotationally positioned nucleosome in vivo". J Biol Chem. 290 (44): 26597–26609. doi:10.1074/jbc.M115.673301. PMC 4646317. PMID 26354431.
- ↑ Jin SG, Pettinga D, Johnson J, Li P, Pfeifer GP (2021). "The major mechanism of melanoma mutations is based on deamination of cytosine in pyrimidine dimers as determined by circle damage sequencing". Science Advances. 7 (31). Bibcode:2021SciA....7.6508J. doi:10.1126/sciadv.abi6508. PMC 8324051. PMID 34330711.
- ↑ Ziegler A, Leffell DJ, Kunala S, Sharma HW, Gailani M, Simon JA, Halperin AJ, Baden HP, Shapiro PE, Bale AE, Brash DE (1993). "Mutation hotspots due to sunlight in the p53 gene of non-melanoma skin cancers". Proceedings of the National Academy of Sciences USA. 90 (9): 4216–20. Bibcode:1993PNAS...90.4216Z. doi:10.1073/pnas.90.9.4216. PMC 46477. PMID 8483937.
- ↑ Mata, Douglas A.; Williams, Erik A.; Sokol, Ethan; Oxnard, Geoffrey R.; Fleischmann, Zoe; Tse, Julie Y.; Decker, Brennan (23 March 2022). "Prevalence of UV Mutational Signatures Among Cutaneous Primary Tumors". JAMA Network Open. 5 (3): e223833. doi:10.1001/jamanetworkopen.2022.3833. PMC 8943639. PMID 35319765.
- ↑ Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR (November 2016). "Mutational signatures associated with tobacco smoking in human cancer". Science. 354 (6312): 618–622. Bibcode:2016Sci...354..618A. doi:10.1126/science.aag0299. PMC 6141049. PMID 27811275.
- ↑ Rogozin IB, Goncearenco A, Lada AG, De S, Yurchenkod V, Nudelman G, Panchenko AR, Cooper DN, Pavlov YI (February 2018). "DNA polymerase η mutational signatures are found in a variety of different types of cancer". Cell Cycle. 17 (3): 348–355. doi:10.1080/15384101.2017.1404208. PMC 5914734. PMID 29139326.
- ↑ Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC (2004). "TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer". IARC Scientific Publications (157): 247–70. PMID 15055300.
- ↑ Pfeifer GP, Hainaut P (2003). "On the origin of G --> T transversions in lung cancer". Mutation Research. 526 (1–2): 39–43. doi:10.1016/s0027-5107(03)00013-7. PMID 12714181.
- 1 2 3 Mosaic, Kat Arney. "The DNA detectives who are hunting the causes of cancer". CNN. Retrieved 2018-09-25.
- ↑ Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. (January 2010). "A comprehensive catalogue of somatic mutations from a human cancer genome". Nature. 463 (7278): 191–6. Bibcode:2010Natur.463..191P. doi:10.1038/nature08658. PMC 3145108. PMID 20016485.
- ↑ Pleasance ED, Stephens PJ, O'Meara S, McBride DJ, Meynert A, Jones D, et al. (January 2010). "A small-cell lung cancer genome with complex signatures of tobacco exposure". Nature. 463 (7278): 184–90. Bibcode:2010Natur.463..184P. doi:10.1038/nature08629. PMC 2880489. PMID 20016488.
- ↑ Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR (January 2013). "Deciphering signatures of mutational processes operative in human cancer". Cell Reports. 3 (1): 246–59. doi:10.1016/j.celrep.2012.12.008. PMC 3588146. PMID 23318258.
- ↑ Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. (August 2013). "Signatures of mutational processes in human cancer". Nature. 500 (7463): 415–21. Bibcode:2013Natur.500..415.. doi:10.1038/nature12477. PMC 3776390. PMID 23945592.
- ↑ Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AW, Boot A, Covington KR, Gordenin DA, Bergstrom E (2018-05-15). "The Repertoire of Mutational Signatures in Human Cancer". bioRxiv 10.1101/322859.
- ↑ Degasperi, Andrea; et al. (21 April 2021). "Substitution mutational signatures in whole-genome–sequenced cancers in the UK population". Science. 376 (6591). doi:10.1126/science.abl9283. PMC 7613262. PMID 35949260.
- ↑ Ledford, Heidi (2022-04-21). "Trove of tumour genomes offers clues to cancer origins". Nature. 604 (7907): 609. Bibcode:2022Natur.604..609L. doi:10.1038/d41586-022-01095-2. PMID 35449305. S2CID 248323597.
- ↑ "Cancer: Huge DNA analysis uncovers new clues". BBC News. 2022-04-21. Retrieved 2022-04-22.